Nile Red Is a Fluorescent Allosteric Substrate of Cytochrome P450 3A4† Jed N
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Exploring the Efficacy of Nile Red in Microplastic Quantification
Letter Cite This: Environ. Sci. Technol. Lett. XXXX, XXX, XXX−XXX pubs.acs.org/journal/estlcu Exploring the Efficacy of Nile Red in Microplastic Quantification: A Costaining Approach Thomas Stanton,*,†,§ Matthew Johnson,† Paul Nathanail,‡ Rachel L. Gomes,§ Teresa Needham,† and Amanda Burson† †School of Geography, University of Nottingham, NG7 2RD Nottingham, United Kingdom ‡Land Quality Management Ltd, University of Nottingham Innovation Park, NG7 2TU Nottingham, United Kingdom §Food, Water, Waste Research Group, Faculty of Engineering, University of Nottingham, NG7 2RD Nottingham, United Kingdom *S Supporting Information ABSTRACT: The presence of microplastic particles (<5 mm) in the environment has generated considerable concern across public, political, and scientific platforms. However, the diversity of microplastics that persist in the environment poses complex analytical challenges for our understanding of their prevalence. The use of the dye Nile red to quantify microplastics is increasingly common. However, its use in microplastic analysis rarely accounts for its affinity with the breadth of particles that occur in environmental samples. Here, we examine Nile red’s ability to stain a variety of microplastic particles and common natural and anthropogenic particles found in environmental samples. To better constrain microplastic estimates using Nile red, we test the coapplication of a second stain that binds to biological material, 4′,6- diamidino-2-phenylindole (DAPI). We test the potential inflation of microplastic estimates using Nile red alone by applying this costaining approach to samples of drinking water and freshwater. The use of Nile red dye alone resulted in a maximum 100% overestimation of microplastic particles. These findings are of particular significance for the public dissemination of findings from an emotive field of study. -

Determining Lipid Content in Embryos Using Nile Red Fluorescence
Laboratory Procedures, PJ Hansen Laboratory - University of Florida Determining Lipid Content in Embryos using Nile Red Fluorescence Luciano Bonilla and Peter J. Hansen Dept. of Animal Sciences, University of Florida The following protocol is derived from the protocol described by Genicot et al. (Theriogenology 63: 1181- 1194, 2005) and is based on the fluorescence emitted by Nile Red when in association with lipid (see Fowler and Greenspan, J Histochem Cytochem 33: 833-836, 1985). Equipments and Reagents o 96 Well plate: BD Falcon®,cat. # 353912, or equivalent o Nile Red: Invitrogen Molecular Probles, cat. # N-1142 o Hoescht 33342: Sigma-Aldrich, cat. # B2261 o ProLong® Gold antifage reagent: Invitrogen Molecular Probes, cat. # P36934 o Fluorescence microscope: Fluorescent microscope configured with excitation 400-500 nm and emission 515 nm Solutions o 100 mM PO4, pH7.4 (Stock solution) To prepare 100 mM PO4 pH 7.4, add sodium phosphate monobasic (1a) to 300 ml sodium phosphate dibasic until the pH reaches 7.4. Approximately, 100 ml of monobasic solution will be added. 1a. 100 mM sodium phosphate monobasic (NaH2PO4.2H2O) 13.8 g NaH2PO4 1 L double–distilled (dd) ddH2O 1b. 100 mM sodium phosphate dibasic (Na2HPO4) 14.2g Na2HPO4.2H2O 1 L ddH2O o 10 mM PBS/PVP (Work solution) Dilute 100 mM PO4, pH 7.4 with 800 ml ddH2O, add 9 g of NaCl and 1.0 g polyvinylpyrollidone (PVP), and bring volume to 1000 ml with water. Check pH. o 4% Paraformaldahyde Fixative - add 500 mL 8% (w/v) paraformaldehyde to 500 mL PBS- PVP (1:1 dilution) o Nile Red Stock Solution (1 mg/mL) - dissolve 25 mg of Nile Red (Invitrogen N-1142) in 25 mL of DMSO; Store at room temperature in the dark indefinitely. -

Spectroscopic Studies of Nile Red in Organic Solvents and Polymers
,loerllof AND Pl-loitll:lltg.IX~ A: CHI'~[ISTiiY ELSEVIER Journal of Photochemistry and Photobiology A: Chemistry 93 ( 199b.~ 57-64 Spectroscopic studies of nile red in organic solvents and polymers Ashim Kumar Dutta, Kenji Kamada, Koji Ohta * Photonic Chemistry Section, Department of Optical Materiolv, Osaka National Research h~stitute, AIST. Ikeda, Osaka 563, Japan Received 20 March 1995; accepted 14 June 1995 Abstract We have studied tile spectroscopic properties of nile red (NR), a highly fluorescent laser dye, ill organic solvents, binary solvent mixtures and polymers. Spectroscopic studies reveal remarkable changes in the absorption and emission band positions and intensities as a function of the polarity of the medium, Such large dmnges have been attributed to the twisted intramolecular charge transfer (TICT) state of the molecule in polar ntedium. Experimental results show that the molecule is sensitive to the polarity of its microenvironment and is an excellent probe for systems presenting restricted geometries. We have incorporated NR into thin films of poly ( melhyl methacrylate) (PMMA) and poly ( vinyl alcohol) (PVA); it is Ibund that the micropolarity in PVA is greater than that in PMMA; in pv ~., the micropolarity corresponds to that of a binary mixture of acetonitrile and water, whereas in PMMA, the micropolarity corresponds ctosely to that of pure acetonitrile. Keywords: Nile red; Twisted intratnolecular charge transfer (TICT) states; Steady state fluorescence; Time-resolved fluorescence; Aggregation-induced dual fluorescence 1. Introduction films [ 13,14], microparticles [ 15,16] and zeolites [ 17,18], have provided information on the role of the topology of these systems in controlling the photophysical properties of Organic polymers have largely replaced conventional the probe molecules. -

Nile Blue Is a Basic Dye of the Oxazine Group, Which Has Been Used in Several Microscopic and Histochemical Techniques (4, 6)
ACTA HISTOCHEM. CYTOCHEM. Vol. 16, No. 3, 1983 LETTER TO THE EDITOR NILE BLUE SULFATE STAINING FOR DEMONSTRATION OF LIPIDS IN FLUORESCENCE MICROSCOPY Nile blue is a basic dye of the oxazine group, which has been used in several microscopic and histochemical techniques (4, 6) . A staining method based on application of Nile blue sulfate was introduced early for the cytochemical demon- stration of lipids (3, 8, 9), showing neutral fats and fatty acids in red and blue, respectively. There is evidence (1, 10) that aqueous solutions of Nile blue sulfate contain the blue cation of the dye, a red oxidation product (Nile red), and the orange-red imino base. According to this view, phospholipids and fatty acids stain blue because they react with the Nile blue cation, meanwhile neutral fats appear in red color. Other observations seem to be in disagreement with this staining mecha- nism (5, 6) . A fluorescence reaction in tissues after staining with Nile blue sulfate and brilliant cresyl blue has been found by Bozzo and Campos Vidal (2). During the course of investigations by using oxazine dyes we have observed that Nile blue sulfate produces a strong fluorescence reaction in lipid droplets. The fat body of Drosophila larvae is a continuous tissue mass which has been chosen as test material because of its abundance in lipidic inclusions. Drosophila hydei testes, surrounded by the fat body, were fixed in 5 % formaldehyde for 1-24 hr, washed, and then immersed in 0.1 mg/ml aqueous Nile blue sulfate (Fluka) for 30 min. After staining, the material was briefly washed and mounted with a drop of water. -
Study of Wood Pitch Emulsions – Interactions with Nile Red and Influence of Ph Master of Science Thesis in Chemistry and Bioscience
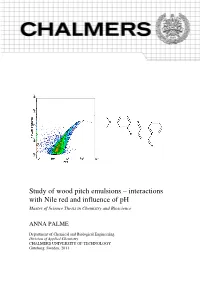
Study of wood pitch emulsions – interactions with Nile red and influence of pH Master of Science Thesis in Chemistry and Bioscience ANNA PALME Department of Chemical and Biological Engineering Division of Applied Chemistry CHALMERS UNIVERSITY OF TECHNOLOGY Göteborg, Sweden, 2011 Study of wood pitch emulsions – interactions with Nile red and influence of pH ANNA PALME Supervisors: Ron Lai and Daniel Persson Examiner: Krister Holmberg Department of Chemical and Biological Engineering Division of Applied Chemistry CHALMERS UNIVERSITY OF TECHNOLOGY Göteborg, Sweden, 2011 Study of wood pitch emulsions – interactions with Nile red and influence of pH. Anna Palme. © Anna Palme, 2011 Department of Chemical and Biological Engineering Division of Applied Chemistry Chalmers University of Technology SE-412 96 Göteborg Sweden Telephone +46 (0)31 772 1000 This work was carried out at Eka Chemicals in Bohus (Sweden). Cover: Left: A density plot from a measurement of Nile red in model pitch emulsion with flow cytometry, showing the red fluorescence intensity versus the forward scattering intensity. Right: The fluorescent probe Nile red. Göteborg, Sweden 2011 Acknowledgements I would like to express my gratitude toward the following people: Ron Lai and Daniel Persson, my supervisors at Eka Chemicals, for their support and engagement in this project. Krister Holmberg for his engagement in the project and for applying a surface chemistry perspective on pitch deposition. The staff at Eka Chemicals for practical assistance in the lab and good company. Anna Palme, Gothenburg, June 2011 Abstract Deposition of wood pitch causes detrimental effects both on the paper and on the paper machine. In this project, it was investigated how a model pitch emulsion is affected by changing the pH from 4 to 8 and back to 4. -

1 High-Throughput Transcriptomic Analysis of Human Primary Hepatocyte Spheroids 1 Exposed to Per- and Polyfluoroalkyl Substances
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.15.341362; this version posted March 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 High-throughput transcriptomic analysis of human primary hepatocyte spheroids 2 exposed to per- and polyfluoroalkyl substances (PFAS) as a platform for relative 3 potency characterization 4 A. Rowan-Carroll1, A. Reardon1, K. Leingartner1, R. Gagné1, A. Williams1, M.J. Meier1, 5 B. Kuo 1, J. Bourdon-Lacombe2, I. Moffat2, R. Carrier2, A. Nong1, L. Lorusso3, S.S. 6 Ferguson4, E. Atlas1 *, C. Yauk5,1* 7 1Environmental Health Science and Research Bureau, Healthy Environments and 8 Consumer Safety Branch (HECSB), Health Canada, 2Water and Air Quality Bureau, 9 HECSB, Health Canada, 3Chemicals and Environmental Health Management Bureau, 10 HECSB, Health Canada, 4US National Institute of Environmental Health Sciences, 11 5Dept. of Biology, University of Ottawa. 12 13 For submission to Toxicological Sciences 14 * To Whom correspondence should be addressed: [email protected] and 15 [email protected] 16 17 Keywords: TempO-Seq, PFAS, liver spheroids, benchmark concentration, new 18 approach methodology, bioactivity exposure ratio 19 This entire data set is publicly available at: at 20 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144775 21 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.15.341362; this version posted March 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. -

Highly Selective Staining and Quantification of Intracellular Lipid Droplets with a Compact Push-Pull Fluorophore Based on Benzothiadiazole S
Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is © The Royal Society of Chemistry 2019 Highly Selective Staining and Quantification of Intracellular Lipid Droplets with a Compact Push-Pull Fluorophore based on Benzothiadiazole S. Israel Suarez,a Caroline C. Warner,b Heather Brown-Harding,a Andrea M. Thooft,b Brett VanVeller,b* and John C. Lukesh IIIa* Table S1. Summary of photophysical data for CBD-Fluor.1 a solvent Polarity value Abs λmax (nm) Em λmax (nm) log ε ΦF Hexane 0.009 427 515 3.60 0.39b HN Toluene 0.099 435 532 3.64 0.30b N Dioxane 0.164 437 546 3.65 0.28b S Dichloromethane 0.309 436 545 3.65 0.26b N Acetonitrile 0.460 437 569 3.64 0.17b Ethanol 0.654 441 578 3.71 0.14c CN Water 1 441 612 3.48 0.005c a Relative solvent polarity scale.2 b Quantum yields determined relative to coumarin 153 in ethanol b (f=0.54) Quantum Yields determined relative to Ru(bpy)3 in 0.5M H2SO4 (f=0.028) Determination of the Limit of Detection for CBD-Fluor Limit of Detection was calculated following the procedure outlined by Wasatch Photonics. https://wasatchphotonics.com/applications/fluorescence-limit-detection/ A series of dilutions from 2.1 µmol to 0.21 nmol of CBD-Fluor were prepared in dioxane and the fluorescence spectra were measured in a cuvette with a 10 mm pathlength. Each sample was excited at 437 nm and the total emission intensity between 447 nm to 700 nm was measured. -

Modification of a Nile Red Staining Method for Microplastics
water Article Modification of a Nile Red Staining Method for Microplastics Analysis: A Nile Red Plate Method Heejun Kang 1,2, Saerom Park 1, Bokjin Lee 1,2, Jaehwan Ahn 1 and Seogku Kim 1,2,* 1 Department of Land, Water and Environment Research, Korea Institute of Civil Engineering and Building Technology (KICT), Goyang 10223, Korea; [email protected] (H.K.); [email protected] (S.P.); [email protected] (B.L.); [email protected] (J.A.) 2 Civil and Environmental Engineering, University of Science and Technology, Daejeon 34113, Korea * Correspondence: [email protected]; Tel.: +82-9100-300 Received: 5 October 2020; Accepted: 17 November 2020; Published: 19 November 2020 Abstract: Recently, environmental pollution from microplastics (MPs) has become a significant reason for increasing the number of studies to develop analysis methods. The Nile red staining method (NR-S), which is staining polymer particles with Nile red (NR) dye, has been widely used for the analysis of MPs in environmental samples. However, this method has several limitations, as it is difficult to stain MPs covered with organic matter residues. In this study, we modified the NR-S method into an NR plate method (NR-P), where the plate is coated with NR instead of staining MPs directly. The optimum concentration of NR solution was obtained (1000 mg/L), and the effectiveness of the NR-P method for the analysis of MPs was assessed using different types (polypropylene, polyethylene, polyethylene terephthalate, and polystyrene), sizes (100–1000 µm), and shapes (sphere, fiber, film, and flake) of plastic materials. -

Safety Data Sheet Packet
Date of Issue: 23 March 2016 SAFETY DATA SHEET PACKET Product Identifier SRM Number: 1934 SRM Name: Fluorescent Dyes for Quantitative Flow Cytometry (Visible Spectral Range) SRM Description: This Standard Reference Material (SRM) is intended for use in assigning fluorescence intensity values to calibration standards for quantitative flow cytometry in the visible spectral range. A unit of SRM 1934 consists of four amber ampoules, each containing 2 mL of a different fluorophore solution or suspension. The solutions include Part A: Fluorescein Solution (60 μM aqueous borate buffer solution); Part B: Nile Red Solution (60 μM acetonitrile solution); Part C: Coumarin 30 Solution (acetonitrile solution); and Part D: APC Suspension (Allophycocyanin fluorescent protein in aqueous buffer solution, 100 µL). SRM 1934 Parts: Part A: Fluorescein Solution Part B: Nile Red Solution Part C: Coumarin 30 Solution Part D: APC Suspension Company Information National Institute of Standards and Technology Standard Reference Materials Program 100 Bureau Drive, Stop 2300 Gaithersburg, Maryland 20899-2300 Telephone: 301-975-2200 Emergency Telephone ChemTrec: FAX: 301-948-3730 1-800-424-9300 (North America) E-mail: [email protected] +1-703-527-3887 (International) Website: http://www.nist.gov/srm This page intentionally left blank. DATE: 23 March 2016 Product Identifier SRM Number: 1934 SRM Name: Fluorescent Dyes for Quantitative Flow Cytometry (Visible Spectral Range) SRM Part: Part A: Fluorescein Solution Under the U.S. Department of Labor, Occupational Safety and Health Administration (OSHA) 29 CFR 1910.1200, this Standard Reference Material (SRM) is NOT classified as a physical hazard or a health hazard, a simple asphyxiant, combustible dust, pyrophoric gas, or hazard not otherwise classified. -

Syntheses and Properties of Water-Soluble Nile Red Derivatives
Syntheses and Properties of Water-Soluble Nile Red Derivatives Jiney Jose and Kevin Burgess* Department of Chemistry, Texas A&M UniVersity, Box 30012, College Station, Texas 77841 [email protected] ReceiVed July 1, 2006 Nile Red (compound A) fluoresces at about 530 nm with good quantum yields in apolar solvents. In more polar ones its fluorescence emission shows a dramatic, and potentially useful, shift to about 640 nm, but its quantum yield is significantly reduced. Further, Nile Red has a very poor solubility in aqueous media. The hypothesis tested in this paper is that Nile Red derivatives that incorporate water-solubilizing groups will tend to fluoresce with good quantum yields in aqueous media, and in the more useful wavelength range around 640 nm. Thus three Nile Red derivatives, 1-3, were prepared. Compound 1 had three hydroxyl groups more than Nile Red, but was surprisingly insoluble in aqueous media. However, the dicarboxylic acid 2 and carboxylic/sulfonic acid derivative 3 showed excellent water solubilities. Spectral data for 2 and 3 showed that they do indeed fluoresce with good quantum yields in the 640 nm region in aqueous media. These properties of compounds 2 and 3 might be useful in the development of fluorescent probes for biotechnology. Introduction development of probes for intracellular imaging, for instance, where emissions become increasingly more detectable with Nile Red, compound A, is a well-known fluorescent dye with 1-3 wavelengths longer than that associated with autofluorescence remarkable solvatochromic properties. In apolar solvents it in the cell (typically above 550 nm). fluoresces with a high quantum yield in the region of 530 nm, Unfortunately, Nile Red itself has very poor solubility in but in polar solvents the quantum yield is dramatically reduced aqueous media,1 so it is not a particularly useful dye for labeling and the emission maximum shows bathochromic shifts of most biomolecules. -

Chemical Visualization of Latent Prints
The author(s) shown below used Federal funds provided by the U.S. Department of Justice and prepared the following final report: Document Title: Chemical Visualization of Latent Prints Author: Evan Durnal Document No.: 238008 Date Received: March 2012 Award Number: 2008-NI-CX-K012 This report has not been published by the U.S. Department of Justice. To provide better customer service, NCJRS has made this Federally- funded grant final report available electronically in addition to traditional paper copies. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Chemical Visualization of Latent Prints Grant # 2008-NI-CX-K012 Final Report For National Institute of Justice Office of Justice Programs U.S. Department of Justice MRI Project No. 110636 July 30, 2010 i Preface The primary author of this report was Mr. Evan Durnal. The report was reviewed by Dr. James Egan and Mr. Tim Lanigan. This document was created in fulfillment of cooperative agreement contract # 2008-NI-CX-K012 Latent Print Chemical Visualization. This document was revised from the DRAFT final report based upon comments provided by The National Institute of Justice review panel. The period of performance for this contract was October 1, 2008 thru July 30, 2010. For technical questions or clarifications regarding this document, contact Dr. James Egan. MIDWEST RESEARCH INSTITUTE //s// Mr. Evan Durnal NIJ Technical Lead Approved: //s// Dr. James Egan NIJ Principal Investigator July 30, 2010 ii Contents Preface ................................................................................................................................. ii Tables ................................................................................................................................. iv Figures............................................................................................................................... -

Location, Dynamics and Solvent Relaxation of a Nile Red-Based
Chemistry and Physics of Lipids 183 (2014) 1–8 Contents lists available at ScienceDirect Chemistry and Physics of Lipids jou rnal homepage: www.elsevier.com/locate/chemphyslip Location, dynamics and solvent relaxation of a nile red-based phase-sensitive fluorescent membrane probe a,1 a,1 a b Roopali Saxena , Sandeep Shrivastava , Sourav Haldar , Andrey S. Klymchenko , a,∗ Amitabha Chattopadhyay a CSIR-Centre for Cellular and Molecular Biology, Uppal Road, Hyderabad 500 007, India b Laboratoire de Biophotonique et Pharmacologie, UMR 7213 CNRS, Université de Strasbourg, Faculté de Pharmacie, 74, Route du Rhin, 67401 Illkirch Cedex, France a r t i c l e i n f o a b s t r a c t Article history: Fluorescent membrane probes offer the advantage of high sensitivity, suitable time resolution, and mul- Received 18 March 2014 tiplicity of measurable parameters, and provide useful information on model and cell membranes. In Received in revised form 22 April 2014 this paper, we have explored the location, dynamics, and solvent relaxation characteristics of a novel Accepted 27 April 2014 Nile Red-based phase-sensitive probe (NR12S). Unlike Nile Red, NR12S enjoys unique orientation and Available online 4 May 2014 location in the membrane, and is localized exclusively in the outer leaflet of the membrane bilayer. By analysis of membrane depth using the parallax approach, we show that the fluorescent group in NR12S is Keywords: localized at the membrane interface, a region characterized by slow solvent relaxation. Our results show Fluorescent membrane probe REES that NR12S exhibits REES (red edge excitation shift), consistent with its interfacial localization.