A 42-Year-Old Female Presented with Asymptomatic Massive Splenomegaly
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Viral Hepatitis with Acute Hemoglobinuria
22 Case Report Viral Hepatitis with Acute Hemoglobinuria Jagabandhu Ghosh1 Joydeep Ghosh2 1 Department of Paediatrics, I.P.G.M.E.R and S.S.K.M Hospital, Kolkata, Address for correspondence Jagabandhu Ghosh, MD (PAED), Ushashi West Bengal, India Housing Society, 245 Vivekananda Road, Kolkata 700006, India 2 Department of Biotechnology, Heritage Institute of Technology, (e-mail: [email protected]). Kolkata, West Bengal, India J Pediatr Infect Dis 2015;10:22–24. Abstract A 7-year-old male child presented with moderate degree fever, yellowish discoloration of eyes and urine. Examination on admission revealed severe anemia, jaundice, hepato- Keywords megaly, and splenomegaly. On the day following admission, the child showed evidence ► hepatitis of blackish discoloration of urine. The diagnosis was established as viral A hepatitis with ► G6PD glucose-6-phosphate dehydrogenase (G6PD) deficiency. The child recovered with ► viral supportive therapy. We suggest that either universal immunization against hepatitis ► hemolysis A, or routine newborn screening for G6PD deficiency, could prevent the serious ► intravascular morbidity or mortality that can occur when these two conditions coexist. Introduction of eyes, and urine for the past 5 days before admission. There was no history of hematemesis, melena, or any other bleed- Acute viral hepatitis A is widely prevalent in India.1 Viral ing, swelling of body, convulsion, and drug ingestion just hepatitis is the leading of acute hepatitis in children in our before present illness or any previous blood transfusion. His country, and hepatitis A is the most common type of viral urine volume was satisfactory. On examination, the child was hepatitis. Hepatitis A does not commonly present with severe deeply icteric, severely anemic, and drowsy. -

An 8-Year-Old Boy with Fever, Splenomegaly, and Pancytopenia
An 8-Year-Old Boy With Fever, Splenomegaly, and Pancytopenia Rachel Offenbacher, MD,a,b Brad Rybinski, BS,b Tuhina Joseph, DO, MS,a,b Nora Rahmani, MD,a,b Thomas Boucher, BS,b Daniel A. Weiser, MDa,b An 8-year-old boy with no significant past medical history presented to his abstract pediatrician with 5 days of fever, diffuse abdominal pain, and pallor. The pediatrician referred the patient to the emergency department (ED), out of concern for possible malignancy. Initial vital signs indicated fever, tachypnea, and tachycardia. Physical examination was significant for marked abdominal distension, hepatosplenomegaly, and abdominal tenderness in the right upper and lower quadrants. Initial laboratory studies were notable for pancytopenia as well as an elevated erythrocyte sedimentation rate and C-reactive protein. aThe Children’s Hospital at Montefiore, Bronx, New York; and bAlbert Einstein College of Medicine, Bronx, New York Computed tomography (CT) of the abdomen and pelvis showed massive splenomegaly. The only significant history of travel was immigration from Dr Offenbacher led the writing of the manuscript, recruited various specialists for writing the Albania 10 months before admission. The patient was admitted to a tertiary manuscript, revised all versions of the manuscript, care children’s hospital and was evaluated by hematology–oncology, and was involved in the care of the patient; infectious disease, genetics, and rheumatology subspecialty teams. Our Mr Rybinski contributed to the writing of the multidisciplinary panel of experts will discuss the evaluation of pancytopenia manuscript and critically revised the manuscript; Dr Joseph contributed to the writing of the manuscript, with apparent multiorgan involvement and the diagnosis and appropriate cared for the patient, and was critically revised all management of a rare disease. -

The Lower Urinary Tract & Male Reproductive System
Chapter 21 Hematopathology Nam Deuk Kim, Ph.D. Pusan National University 1 Contents I. Red blood cells II. Hemostasis III. White blood cells IV. Disorders of the lymphopoietic system V. Spleen VI. Thymus 2 THE HEMATOPOIETIC SYSTEM Composition of Human Blood Blood • Transports oxygen, nutrients, hormones, leukocytes (white cells), red cells, platelets and antibodies to tissues in the body and carbon dioxide and other waste products of cell metabolism to the excretory organs of the body • Volume of blood: Represents about 8% of total body weight • approximately 5 quarts, but it varies according to size of individual (5 liters in women; 5.5 liters in men) • Almost half of the blood consists of cellular elements suspended in plasma (viscous fluid) 3 4 Hemopoiesis Cellular differentiation and maturation of the lymphoid and myeloid components of the hematopoietic system. Only the precursor cells (blasts and maturing cells) are identifiable by light microscopic evaluation of the bone marrow. BFU = burst-forming unit; CFU = colony-forming unit (Ba = basophils; E = erythroid; Eo = eosinophils; G = polymorphonuclear leukocytes; GM = granulocyte-monocyte; M = monocyte/macrophages; Meg = megakaryocytic); EPO = erythropoietin; Gm-CSF = granulocyte-macrophage colony-stimulating factor; IL = interleukin; NK = natural killer; SCF = stem cell factor; TPO = thrombopoietin. 5 Composition of Human Blood • All blood cells arise from precursor cells within the bone marrow, called stem cells • These undergo further differentiation to form red cells, white cells, -
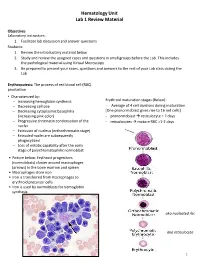
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Hemophagocytic Lymphohistiocytosis in a Child with Sickle Cell Disease
Hematology & Transfusion International Journal Case Report Open Access Hemophagocytic lymphohistiocytosis in a child with sickle cell disease Keywords: hemophagocytic lymphohistiocytosis, FHL, EBV, hemoglobin S, sickle cell disease, X-ray Volume 6 Issue 5 - 2018 Introduction Walaa Shoman,1 Yasmine El Chazli,2 Asmaa Elsharkawy,2 Neveen Mikhael,3 Akram Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening deghaidy,3 Abeer Al-Battashi,4 Yasser Wali2 hyper-inflammatory syndrome which represents the extreme end of 1Department of Pediatrics, Immunology/Rheumatology unit, a severe uncontrolled hyperinflammatory reaction that can occur in Alexandria University, Egypt many underlying conditions.1 HLH can be divided into primary and 2Department of Pediatrics, Hematology/Oncology unit, secondary HLH. Primary HLH is caused by gene mutation, either at Alexandria University, Egypt 3 one of the Familial HLH (FHL) loci or in a gene responsible for one Department of Clinical pathology, Alexandria University, Egypt 4Department of Child Health, Royal Hospital, Oman of several immunodeficiency syndromes.2 The most common form of secondary HLH is infection associated HLH. Infectious triggers Correspondence: Yasser Wali, Department of Pediatrics, include viruses (as EBV, cytomegalovirus, HHV8, HIV), bacteria (as Hematology/Oncology unit, Alexandria University, Egypt, mycobacteria, mycoplasma), parasites (as leishmania, plasmodium), Email [email protected] 3,4 and fungi (as candida, cryptococcus). Sickle cell disease (SCD) Received: September 02, 2018 | Published: September 27, and its variants are genetic disorders resulting from the presence 2018 of a mutated form of hemoglobin, hemoglobin S (HbS), which can be detected by hemoglobin electrophoresis. SCD is suggested by a high-grade unremitting fever, grunting, marked pallor, jaundice, the typical clinical picture of chronic hemolytic anemia and vaso- and marked re-enlargement of both liver and spleen despite improved occlusive crisis.5,6 We present a case of HLH in an Egyptian boy who chest condition and radiogram. -

Case Report Gastric Volvulus Due to Splenomegaly - a Rare Entity
Case Report Gastric Volvulus Due to Splenomegaly - A rare Entity. Ashok Mhaske Department of Surgery,People’s College of Medical Sciences and Research Centre, Bhanpur, Bhopal. 462010 , (M.P.) Abstract: Gastric volvulus is a rare condition which typically presents with intermittent episodes of abdominal pain. The volvulus occurs around an axis made by two fixed points, organo-axial or mesentero-axial. Increased pressure within the hernial sac associated with gastric distension can lead to ischemia and perforation. Acute obstruction of a gastric volvulus is thus a surgical emergency.We present an unusual case of gastric volvulus secondary to tropical splenomegaly. Tropical splenomegaly precipitating gastric volvulus is not documented till date. Key Words: Gastric volvulus, Tropical splenomegaly. Introduction: to general practitioner but as symptoms persisted she Gastric volvulus is abnormal rotation of the was referred to our tertiary care centre for expert stomach of more than 180 degrees. It can cause closed opinion and management. loop obstrcution and at times can result in On examination, vital parameters and hydration was adequate with marked tenderness in incarceration and strangulation. Berti first described epigastric region with vague lump. Her routine gastric volvulus in 1866; to date, it remains a rare investigations were inconclusive but she did partially clinical entity. Berg performed the first successful responded to antispasmodic and antacid drugs. operation on a patient with gastric volvulus in 1896. Ultrasound of abdomen and Barium studies Borchardt (1904) described the classic triad of severe revealed gastric volvulus (Fig.1 & 2) with epigastric pain, retching without vomiting and splenomegaly for which she was taken up for inability to pass a nasogastric tube. -

Hereditary Spherocytosis with Gilbert's Syndrome
IOSR Journal of Dental and Medical Sciences (IOSR-JDMS) e-ISSN: 2279-0853, p-ISSN: 2279-0861.Volume 20, Issue 4 Ser.6 (April. 2021), PP 25-28 www.iosrjournals.org Hereditary Spherocytosis with Gilbert’s Syndrome – a case report. Dr Deepan Panneerselvam Intern, Department of Internal Medicine. Dr Thabuna Sivaprakasam Intern, Department of Internal Medicine. Dr Raja sekar PGY2 Internal Medicine Resident. Dr Govindarajulu Ethirajulu Chief, Department of Internal Medicine. Government Kilpauk Medical College and Hospital. Abstract: A 23 year old previously diagnosed female with Gilbert’s syndrome on treatment with Prednisone, presented with a lower respiratory tract infection for the past 4 days with additional complaints of significant lethargy and fatigue, palpitation and intermittent yellowish discolouration of sclera since 9 years old. The patient’s mother and elder sister also had similar history of intermittent jaundice. Routine investigations of the patient revealed normal values except a hemoglobin level of 6.0 g/dl, HCT of 19.5%, MCV of 75.6 fL, serum total bilirubin of 6.3 mg/dl ( Direct: 1.3mgdl, Indirect: 5.0 mg/dl ). This made us suspect another basic hematologic abnormality that contributed to such elevated indirect bilirubin levels. Further investigations revealed a peripheral smear with moderate microcytic hypochromic anemia, anisopoikilocytosis, leucopenia and a reticulocyte count of 2.3%, negative Direct coombs test and a serum LDH of 345U/L. Ultrasound sound abdomen revealed multiple calculi in gall bladder largest measuring 3mm and splenomegaly. Peripheral smear was repeated only to reveal a dimorphic blood picture with eosinophilia, normocytic normochromic anemia with a few fragmented RBCs and spherocytes. -

FDA CVM Comprehensive ADE Report Listing for Afoxolaner
CVM ADE Comprehensive Clinical Detail Report Listing Cumulative Date Range : 04-Sep-2013 -thru- 31-Jul-2018 Included 1932a cases = : True Included Medicated Feed cases = : False DRUG: AFOXOLANER Species: Cat Route of Administration: oral Sign: VOMITING, Number of times reported: 4 Sign: HYPERACTIVITY, Number of times reported: 3 Sign: ITCHING, Number of times reported: 3 Sign: LETHARGY, Number of times reported: 3 Sign: PRURITUS, Number of times reported: 3 Sign: ACCIDENTAL EXPOSURE, Number of times reported: 2 Sign: ANOREXIA, Number of times reported: 2 Sign: LABOURED BREATHING, Number of times reported: 2 Sign: NOT EATING, Number of times reported: 2 Sign: PANTING, Number of times reported: 2 Sign: SEIZURE NOS, Number of times reported: 2 Sign: ABNORMAL TEST RESULT, Number of times reported: 1 Sign: AGITATION, Number of times reported: 1 Sign: ALOPECIA, Number of times reported: 1 Sign: ANAEMIA NOS, Number of times reported: 1 Sign: ATAXIA, Number of times reported: 1 Sign: CERVICAL VENTROFLEXION, Number of times reported: 1 Sign: CONSTIPATION, Number of times reported: 1 Sign: DECREASED CHOLESTEROL (TOTAL), Number of times reported: 1 Sign: ELEVATED ALT, Number of times reported: 1 Sign: ELEVATED AST, Number of times reported: 1 Sign: ELEVATED BUN, Number of times reported: 1 Sign: ELEVATED CREATINE-KINASE (CK), Number of times reported: 1 Sign: ELEVATED CREATININE, Number of times reported: 1 Sign: ELEVATED TOTAL BILIRUBIN, Number of times reported: 1 Sign: EXCESSIVE LICKING AND/OR GROOMING, Number of times reported: 1 Sign: FEVER, -

Laboratory Approach to Anemia Laboratory Approach to Anemia
DOI: 10.5772/intechopen.70359 Provisional chapter Chapter 12 Laboratory Approach to Anemia Laboratory Approach to Anemia Ebru Dündar Yenilmez and Abdullah Tuli Ebru Dündar Yenilmez and Abdullah Tuli Additional information is available at the end of the chapter Additional information is available at the end of the chapter http://dx.doi.org/10.5772/intechopen.70359 Abstract Anemia is a major cause of morbidity and mortality worldwide and can be defined as a decreased quantity of circulating red blood cells (RBCs). The epidemiological studies suggested that one-third of the world’s population is affected with anemia. Anemia is not a disease, but it is instead the sign of an underlying basic pathological process. However, the sign may function as a compass in the search for the cause. Therefore, the prediag- nosis revealed by thorough investigation of this sign should be supported by laboratory parameters according to the underlying pathological process. We expect that this review will provide guidance to clinicians with findings and laboratory tests that can be followed from the initial stage in the anemia search. Keywords: anemia, complete blood count, red blood cell indices, reticulocyte 1. Introduction Anemia, the meaning of which in Greek is “without blood,” is a relatively common sign and symptom of various medical conditions. Anemia is defined as a significant decrease in the count of total erythrocyte [red blood cell (RBC)] mass, although this definition is rarely used in clinical settings. According to the World Health Organization, anemia is a condition in which the number of red blood cells (RBCs, and consequently their oxygen-carrying capacity) is insufficient to meet the body’s physiologic needs [1, 2]. -

Anemic Syndrome and White Blood Cells Disorders
27. 11. 2020 Anemic syndrome and white blood cells disorders Kristína Repová, M.D., PhD. [email protected] Institute of Pathophysiology, Faculty of Medicine, Bratislava Prepared exclusively for the purposes of distance education at the Faculty of Medicine, Comenius University in Bratislava in 2020/21 Hematopoeisis • Hematopoietic organs: • Bone marrow: • forming of erythrocytes, granulocytes, monocytes, thrombocytes, partially lymphocytes • Thymus: • forming of T-lymphocytes • Lymphatic nodes, tonsils, spleen: • forming of B-lymphocytes lymphoid multipotent stem cell pluripotent progenitor cell precursor cell stem cell myleoid multipotent stem cell 1 27. 11. 2020 Hematopoeisis 3 Pluripotent hematopoietic stem cell (self-renewal) Myeloid multipotent Lymphoid multipotent stem cell stem cell Megacaryocyte and Granulocyte and T-cell and NK B-cell erythroid progenitor Macrophage progenitor cell progenitor progenitor Megacaryocyte Erythrocyte Granulocyte Monocyte progenitor progenitor progenitor progenitor (CFU-Meg) (CFU-E) (CFU-G) (CFU-M) Myeloblast NK-cell Proerythroblast Monoblast Lymphoblast Lymphoblast Promyelocyte Megacaryoblast Erythroblast Myelocyte Promonocyte Prolymphocyte Prolymphocyte Megacaryocyte Reticulocyte Metamyelocyte Monocyte T-cell B-cell Thrombocyte Erythrocyte Band cell Basophil Eosinophil Macrophage Dendritic cell Neutrophil 2 27. 11. 2020 I. Disorders of red blood cells II. Disorders of white blood cells III. Myeloproliferative and lymphoproliferative disorders I. Disorders of red blood cells 1. Anemia 2. -

Hemolytic Anemia Presenting As Hemoglobinuria from Intentional Paradichlorobenzene Mothball Ingestion
Hemolytic Anemia from Intentional ParadichlorobenzeneCASE Mothball REPORT Ingestion Hemolytic Anemia Presenting as Hemoglobinuria from Intentional Paradichlorobenzene Mothball Ingestion Mary Ondinee U. Manalo,1 Cherie Grace G. Quingking2 and Carissa Paz C.Dioquino2 1Department of Medicine, College of Medicine and Philippine General Hospital, University of the Philippines Manila 2National Poison Management and Control Center, University of the Philippines-Philippine General Hospital less acutely toxic of the two. However, like naphthalene, it has also been known to induce hemolytic anemia because it possesses one benzene ring.1 We report a case of a man who intentionally ingested three mothballs made of paradicholorobenzene who experienced severe hemolytic anemia that necessitated blood transfusion. Case A 24-year-old male inmate from Manila was admitted for persistent vomiting three days after ingestion of three crushed mothballs. Three days prior to admission, the patient intentionally swallowed three crushed mothballs. After an hour, he experienced nausea and vague abdominal pain. A day prior to admission, he presented with persistent vomiting and passed out dark stools. Eight hours prior to admission, Introduction vomiting became more frequent and was now associated Mothballs could be made of naphthalene, with coffee-ground material. Abdominal pain became more paradichlorobenzene, or camphor. Differentiation among intense as well. He was then brought to the Emergency mothballs is difficult because they may have similar odors Room of the Philippine General Hospital for consultation. and are all white, crystalline solids at room temperature. The patient presented at the emergency room awake but Paradichlorobenzene is commonly found here in the weak-looking, tachypneic but with otherwise stable vital Philippines as a component of toilet deodorant blocks, but signs. -

432 Medicine Team Leaders 3:C
Reviewed By: Done By: Naif Aleid Abdulhameed Alsaawy Abdulrahman AlZahrani COLOR GUIDE: • Females' Notes • Males' Notes • Important • Additional 432MedicineTeam Anemia Objectives: (not given) General Outline Microcytic-hypochromic anaemias Normocytic-normochromic Morphology anaemias Macrocytic- normochromic anaemias Dyshaemopoietic anaemia Classification (deficiencies impairing development) Decrease red cell production Hypoproliferative anaemias Anemia (BM failure ): Haemolytic anaemia Etioilogy Acute post haemorrhagic anaemia DilutIonal anaemia 1 432MedicineTeam Anemia Introduction: Blood formation: 1. Intrauterine life: Blood cells are formed in the liver and spleen up to the fifth months. After 5th month: bone marrow shares in the formation of these cells. 2. After birth: Formation of these cells will be restricted to the bone morrow. 3. Adulthood The active bone morrow will be restricted to axial skeleton: flat bones, vertebrae, ribs, sternum and ilia. Some extension to the proximal ends of long bone mainly femur. Extramedullary haemopoiesis: When demand for blood formation is increased the active red BM extends into the shafts of the long bones. The spleen and liver will regain their ability to produce blood elements when bone morrow is affected by some diseases. Blood Constituents: Organelles (formed elements): o RBCs o WBCs o Thrombocytes Fluid components: plasma in which the above elements are suspended and contain fibrinogen. 2 432MedicineTeam Anemia Erythrocytopoiesis Normal erythrocytopoiesis depends on: 1. Healthy BM: normal stem cells and architecture. 2. Regulatory hormones: GPO, Androgen thyroxin, cortisol and ACT. 3. Nutritional elements: Protein (high biological value), 4. Minerals (Iron, copper, zinc, selenium) 5. Vitamins (B, Folic acid, vit. C) Anaemia: Reduction of O2 carrying capacity of the blood with inadequate O2 supply to tissues.