A Practical Chiral Auxiliary for Asymmetric Synthesis** Marvin R
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Chiral Auxiliaries and Optical Resolving Agents
Chiral Auxiliaries and Optical Resolving Agents Most bioactive substances are optically active. For instance, if This brochure introduces a variety of chiral auxiliaries and a substance is synthesized as a racemic compound, its optical resolving agents. We hope that it will be useful for your enantiomer may show no activity or even undesired bioactivity. research of the synthesis of optically active compounds. Thus, methods to gain enantiopure compounds have been Additionally, TCI has some brochures introducing chiral developed. When synthesizing enantiopure compounds, the compounds for the chiral pool method in “Chiral Building Blocks”, methods are roughly divided into three methods. “Terpenes”, “Amino Acids” and other brochures. Sugar derivatives are also introduced in a catalog, “Reagents for Glyco Chemistry Chiral pool method: & Biology”, and category pages of sugar chains. Furthermore, The method using an easily available chiral compound as a TCI has many kinds of catalysts for asymmetric synthesis and starting material like an amino acid or sugar. introduce them in brochures such as “Asymmetric Synthesis” and Asymmetric synthesis: “Asymmetric Organocatalysts”, and other contents. The method to introduce an asymmetric point to compounds You can search our information through “asymmetric synthesis” without an asymmetric point. Syntheses using achiral as a keyword. auxiliaries are included here. Optical resolution: The method to separate a racemic compound into two ● Reactions with Chiral Auxiliaries enantiomers. The direct method using a chiral column and One of the most famous named reactions using chiral auxiliaries1) the indirect method to separate two enantiomers using is the Evans aldol reaction.2) This reaction is quite useful because optical resolving agents to convert into diastereomers are this reaction can efficiently introduce two asymmetric carbons into examples. -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Chapter 8. Chiral Catalysts José M
Chapter 8. Chiral Catalysts José M. Fraile, José I. García, José A. Mayoral 1. The Origin of Enantioselectivity in Catalytic Processes: the Nanoscale of Enantioselective Catalysis. Enantiomerically pure compounds are extremely important in fields such as medicine and pharmacy, nutrition, or materials with optical properties. Among the different methods to obtain enantiomerically pure compounds, asymmetric catalysis1 is probably the most interesting and challenging, in fact one single molecule of chiral catalyst can transfer its chiral information to thousands or even millions of new chiral molecules. Enantioselective reactions are the result of the competition between different possible diastereomeric reaction pathways, through diastereomeric transition states, when the prochiral substrate complexed to the chiral catalyst reacts with the corresponding reagent. The efficiency of the chirality transfer, measured as enantiomeric excess [% ee = (R−S)/(R+S) × 100], depends on electronic and steric factors in a very subtle form. A simple calculation shows that differences in energy of only 2 kcal/mol between these transition states are enough to obtain more than 90% ee, and small changes in any of the participants in the catalytic process can modify significantly this difference in energy. Those modifications may occur in the near environment of the catalytic centre, at less than 1 nm scale, but also at longer distances in the catalyst, substrate, reagent, solvent, or support in the case of immobilized catalysts. This is the reason because asymmetric -

APPLICATIONS in ASYMMETRIC SYNTHESIS Carlos
324 SYNTHESIS OF OCTAHYDROBENZO - 1, 2,3 - DIAZAPHOSPHOLIDINE - 2 - OXIDES AND THEIR DERIVATIVES: APPLICATIONS IN ASYMMETRIC SYNTHESIS DOI: http://dx.medra.org/ 10.17374/targets.2020.23.324 Carlos Cruz - Hernández a , José M. Landeros a , Eusebio Juaristi * a,b a Departamento de Química, Centro de Investigación y de E studios Avanzados, Avenida IPN 2508, 07360 Ciudad de México, Mexico b El Colegio Nacional, Luis González Obregón 23, Centro Histórico, 06020 Ciudad de México, Mexico (e - mail: [email protected]; [email protected]) Abstract. This chapter outlines recent efforts devoted to the synthesis of heterocycles that include the octahydrobenzo - 1,3,2 - diazaphospholidine - 2 - oxide fragment, as well as their application in asymmetri c synthesis. The first part of this review provides a brief discussion of the general structural characteristics of this phosphorus - containing heterocyclic scaffold. The second part describes the synthetic paths that were undertaken to synthesize the desir ed heterocycles , as well as some relevant considerations pertaining the spectroscopic characterization of the phosphorus - containing heterocycles of interest . The third part provides several illustrative examples where the novel chiral heterocycles were employed in ena ntioselective synthesis. The new phosphorus - containing heterocycles proved useful : i) as chiral auxiliaries in nucleophilic addition reactions, as well as as imine activators in electrophilic addition reactions; ii ) as c hiral ligands i n nucleophilic allylation and crotonylation of prochiral aldehydes , and iii) as c hiral organocatalysts in enantioselective aldol, Michael, and cascade reactions. Contents 1. Introduction 2. Synthesis of the octahydrobenzo - 1,3,2 - diazaphospho lidine - 2 - oxide s 2.1 . Conformational and configurational assignments 3 . -

Application of Chiral Sulfoxides in Asymmetric Synthesis
MOJ Bioorganic & Organic Chemistry Review Article Open Access Application of chiral sulfoxides in asymmetric synthesis Abstract Volume 2 Issue 2 - 2018 Chiral sulfoxides are used as a toolbox for the synthesis of enantiomeric/diastereomeric compounds, which are used as precursors for the pharmaceutically/chemically Ganapathy Subramanian Sankaran,1 important molecules. The current review focuses on applying these chiral sulfoxides Srinivasan Arumugan,2 Sivaraman towards the synthesis of the compounds having stereogenic center. In general, the 3 stereogenic center induced by the sulfoxide is able to direct the stereochemistry of Balasubramaniam 1University of Massachusetts Medical School, USA further transformation necessary to complete the total synthesis of bioactive molecules. 2Department of Science and Humanity (Chemistry), Karunya The nature of the reactive conformation of the sulfoxide is strongly dependent on the Institute of Technology and Sciences, India nature of the substituents at C-α and/or C-β. 3Indian Institute of Technology Madras, Chennai, India Correspondence: Sivaraman Balasubramaniam, Senior Research Scientist, Indian Institute of Technology Madras, Chennai, India, Tel +9177 1880 5113, Email [email protected] Received: March 07, 2018 | Published: March 29, 2018 Introduction occurred in a further oxidation step of one of the sulfinyl enantiomer to sulfone.13 The titanium-binaphthol complex catalyzes not only the Over the last three decades, the sulfinyl group has received asymmetric oxidation but also the kinetic -

Asymmetric Synthesis
Chapter 45 — Asymmetric synthesis - Pure enantiomers from Nature: the chiral pool and chiral induction - Asymmetric synthesis: chiral auxiliaries Enolate alkylation Aldol reaction - Enantiomeric excess (ee) - Asymmetric synthesis: chiral reagents and catalysts CBS reagent for chiral reductions Sharpless asymmetric epoxidation Synthesizing pure enantiomers starting from Nature’s chiral pool AcO OH HO B O O HO – O O HO + O N OH H3N OH HO OH O H … AcO HO OH O OH Sulcatol - insect pheremone HO Synthesis from the chiral pool — chiral induction turns one stereocenter into many 40 steps Me HO H Me H O O OH O O OBn O * * Only one H O HO chiral reagent H H Me OBn Me Deoxyribose Fragment of Brevetoxin B Asymmetric synthesis 1. Producing a new stereogenic centre on an achiral molecule makes two enantiomeric transition states of equal energy… and therefore two enantiomeric products in equal amounts. O O 1 1 Nu R R2 R R2 Nu E OH OH 1 O 1 Nu R R Nu R2 R2 1 R R2 O Ph OH HO Ph PhLi + N N N Asymmetric synthesis 2. O 1 R R* Nu When there is an existing O chiral centre, the two possible TS’s are diastereomeric and E 1 Nu R R* can be of different energy. Thus one isomer of the new stereogenic centre can be OH O OH produced in a larger amount. 1 1 R Nu Nu R *R 1 R* R R* HO Ph O O O Ph OH PhLi PhLi Ph Ph N N N N N Ph N N N N N OH Ph O O O Ph OH Ph A removable chiral centre… synthesis with chiral auxiliaries O ??? O R R 1) Add a chiral 3) Remove the auxiliary chiral auxiliary O O R* R* 2) Add the new stereocentre via chiral induction Enantiopure oxazolidinones as chiral auxiliaries 1. -

A Sustainable, Two-Enzyme, One-Pot Procedure
A Sustainable, Two-Enzyme, One-Pot Procedure for the Synthesis of Enantiomerically Pure α-Hydroxy Acids Andrzej Chmura Cover picture: Manihot esculenta (cassava), source: http://www.hear.org/starr/images/image/?q=090618-1234&o=plants SEM photograph of an Me HnL CLEA (author: Dr. Rob Schoevaart, CLEA Technologies, Delft, The Netherlands) Cover design by Andrzej Chmura A Sustainable, Two-Enzyme, One-Pot Procedure for the Synthesis of Enantiomerically Pure α-Hydroxy Acids PROEFSCHRIFT ter verkrijging van de graad van doctor aan de Technische Universiteit Delft, op gezag van de Rector Magnificus prof.ir. K.C.A.M. Luyben, voorzitter van het College voor Promoties, in het openbaar te verdedigen op dinsdag 7 december 2010 om 12.30 uur door Andrzej CHMURA Magister inŜynier in Chemical Technology, Politechnika Wrocławska, Wrocław, Polen en Ingenieur in Chemistry, Hogeschool Zeeland, Vlissingen, Nederland geboren te Łańcut, Polen Dit proefschrift is goedgekeurd door de promotor: Prof. dr. R.A. Sheldon Copromotor: Dr. ir. F. van Rantwijk Samenstelling promotiecommissie: Rector Magnificus Voorzitter Prof. dr. R.A. Sheldon Technische Universiteit Delft, promotor Dr. ir. F. van Rantwijk Technische Universiteit Delft, copromotor Prof. dr. I.W.C.E. Arends Technische Universiteit Delft Prof. dr. W.R. Hagen Technische Universiteit Delft em. Prof. dr. A.P.G. Kieboom Universiteit Leiden Prof. dr. A. Stolz Universität Stuttgart Prof. dr. V. Švedas Lomonosov Moscow State University Prof. dr. J.J. Heijnen Technische Universiteit Delft, reserve lid The research described in this thesis was financially supported by The Netherlands Research Council NWO under the CERC3 programme and by COST action D25. ISBN/EAN: 978-90-9025856-0 Copyright © 2010 by Andrzej Chmura All rights reserved. -

Stereoselective Synthesis of Chiral
S S symmetry Article Stereoselective Synthesis of Chiral α-SCF3-β-Ketoesters FeaturingArticle a Quaternary Stereocenter Stereoselective Synthesis of Chiral α-SCF3-β-Ketoesters Monica Fiorenza Boselli, Chiara Faverio, Elisabetta Massolo, Laura Raimondi, Alessandra Puglisi andFeaturing Maurizio Benaglia a Quaternary * Stereocenter Monica Fiorenza Boselli, ChiaraDipartimento Faverio, diElisabetta Chimica, UniversitMassolo,à Lauradegli Studi Raimondi, di Milano, Alessandra Via Golgi 19,Puglisi I-20133 and Milano, Maurizio Italy; Benaglia * monicafi[email protected] (M.F.B.); [email protected] (C.F.); [email protected] (E.M.); [email protected] (L.R.); [email protected] (A.P.) *DipartimentoCorrespondence: di Chimica, [email protected]; Università degli Studi di Milano, Tel.: Via +39-02-5031-4171; Golgi 19, I-20133 Milano, Fax: +39-02-5031-4159 Italy; [email protected] (M.F.B.); [email protected] (C.F.); [email protected] (E.M.); Abstract:[email protected] development (L.R.); of [email protected] new and efficient methods, (A.P.) reagents, and catalysts for the introduction * Correspondence: [email protected]; Tel.: +39-02-5031-4171; Fax: +39-02-5031-4159 of fluorine atoms or fluorinated moieties in molecular scaffolds has become a topic of paramount importanceAbstract: The in development organic synthesis. of new and In thisefficient framework, methods, thereagents, incorporation and catalysts of thefor SCFthe introduc-3 group into organic moleculetion of fluorine has oftenatoms ledor fluorinated to beneficial moieties effects in molecular on the drug’s scaffolds metabolic has become stability a topic andof para- bioavailability. Heremount we importance report ourin organic studies synthesis. -
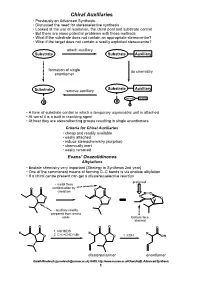
Chiral Auxiliaries • Previously on Advanced Synthesis
Chiral Auxiliaries • Previously on Advanced Synthesis..... • Discussed the need for stereoselective synthesis • Looked at the use of resolution, the chiral pool and substrate control • But there are some potential problems with those methods • What if the substrate does not contain an appropriate stereocentre? • What if the target does not contain a readily exploited stereocentre? attach auxiliary Substrate Substrate Auxiliary formation of single do chemistry enantiomer Substrate Auxiliary Substrate remove auxiliary Substrate Auxiliary R R R • A form of substrate control in which a temporary asymmetric unit is attached • At worst it is a built in resolving agent • At best they are stereodirecting groups resulting in single enantiomers Criteria for Chiral Auxiliaries • cheap and readily available • easily attached • induce stereochemistry (surprise) • chemically inert • easily removed Evans' Oxazolidinones Alkylations • Enolate chemistry very important (Strategy in Synthesis 2nd year) • One of the commonest means of forming C–C bonds is via enolate alkylation • If a chiral centre present can get a diastereoselective reaction preferred • metal fixes M O O conformation by O chelation O M O N O O O N O N H • auxiliary readily prepared from amino acids Bottom face blocked O O O O O 1. NaHMDS R R R N O 2. CH2=CHCH2Br N O 1. KOH OH diastereoisomer enantiomer Gareth Rowlands ([email protected]) Ar402, http://www.sussex.ac.uk/Users/kafj6, Advanced Synthesis 1 • The aldol reaction is an extremely valuable means of introducing stereochemistry -

Synthesis and Characterization of Chiral Monomers and Polymers Syed Aziz Ashraf University of Wollongong, [email protected]
University of Wollongong Research Online University of Wollongong Thesis Collection University of Wollongong Thesis Collections 1997 Synthesis and characterization of chiral monomers and polymers Syed Aziz Ashraf University of Wollongong, [email protected] Recommended Citation Ashraf, Syed Aziz, Synthesis and characterization of chiral monomers and polymers, Doctor of Philosophy thesis, Department of Chemistry, University of Wollongong, 1997. http://ro.uow.edu.au/theses/1187 Research Online is the open access institutional repository for the University of Wollongong. For further information contact the UOW Library: [email protected] SYNTHESIS AND CHARACTERIZATION OF CHIRAL MONOMERS AND POLYMERS A thesis submitted in fulfilment of the requirements for the award of the degree DOCTOR OF PHILOSOPHY from UNIVERSITY OF WOLLONGONG by SYED AZIZ ASHRAF M.Sc DEPARTMENT OF CHEMISTRY June 1997 DECLARATION This is to certify that the work described in this thesis has not been submitted for a higher degree at any other University or institution. Syed Aziz Ashraf I ACKNOWLEDGMENTS I wish to express my sincere gratitude to my supervisors Professor Gordon G. Wallace, Professor Leon A.P. Kane-Maguire and Associate Professor Stephen G. Pyne for their enthusiastic supervision and encouragements throughout the completion of my thesis. I greatly appreciate the help and discussion of Professor Alan G. MacDiarmid of University of Pennsylvania, Philadelphia, USA, where I have carried out some of the experimental work of Chapter 3. The helps and friendships of staff and my fellow colleagues in Chemistry department are also greatly appreciated, especially Chee O. Too, Mir Reza Majidi, Zhao Hujin, Richard John, Ian Norris, Trevor Lewis and Norm Barisi, for their support and fruitful discussion. -
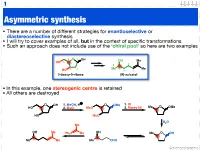
Asymmetric Synthesis
1 Asymmetric synthesis • There are a number of different strategies for enantioselective or diastereoselective synthesis • I will try to cover examples of all, but in the context of specific transformations • Such an approach does not include use of the ‘chiral pool’ so here are two examples 4 O 1 OH OH Me HO 5 5 2 3 2 Me 4 Me HO 3 1 2-deoxy-D-ribose (R)-sulcatol • In this example, one stereogenic centre is retained • All others are destroyed O OH 1. MeOH, H O OMe 1. KI HO 2. MsCl MsO 2. Raney Ni Me O OMe HO MsO H2O Me OH Me OH Ph3P Me Me O OH Me Me Me CHO Advanced organic 2 ‘Chiral pool’ II Me Me remove OH Me Me stereogenic 3 steps O O centre HO 2 4 OH PCC 3 O OH O O 6 OH HO 1 O 5 OTBDPS OTBDPS D-mannose BnO O BnO O addition of 1. NaBH protecting groups overall retention of 4 stereochemistry 2. Tf2O stereoselective Me 1. TBAF Me Me reduction Me 2. PCC Me Me O O O 3. Ph P=CHCHO NaN O 3 O 3 O N3 N3 OTf OTBDPS OTBDPS BnO O CHO BnO O BnO O Pd / C hydrogenolysis of benzyl (Bn) group & H reductive amination of resultant 2 aldehyde Me HO two step reversal of Me O 2 1 stereogenic centre reduction 1. H2, Pd / C, H of alkene & H H 3 O N 2. TFAA azide HO 4 N followed by reductive H 6 amination BnO O 5 H HO swainsonine • In this example three stereogenic centres are retained • One stereogenic centre undergoes multiple inversion -- but overall it is retained Advanced organic 3 Stereoselective reactions of alkenes • Alkenes are versatile functional groups that, as we shall see, present plenty of scope for the introduction of stereochemistry • Hydroboration permits the selective introduction of boron (surprise), which itself can undergo a wide-range of stereospecific reactions Substrate control Me H H Me H H Me 1. -

7-Chapter 1.Pdf
Chapter 1 1.1.1 Chirality: Definition and its types: Until half a century ago, it was assumed that the forces of nature were symmetric and that they did not distinguish between right and left, between image and mirror image. The discovery of the violation of parity in 1956 was more than a sensation, for some it was a shock. It implied that the universe displays handedness, or chirality, and that it is fundamentally asymmetric.1 The idea of chirality has been known in chemistry since chiral chemistry got impetus from pioneering work by Louis Pasteur, a French chemist and biologist, who physically separated the two isomers of sodium ammonium tartrate in 1848 although it would be nearly a hundred years before chemists began using this term. In fact, in the first edition of Eliel's "Stereochemistry of Carbon Compounds" in 1962, the word chiral is not mentioned.2 The definition of chirality was first given by Lord Kelvin in May 1893, during a conference of the Oxford University Junior Scientific Club: “I call any geometrical figure, or group of points, chiral, and say it has chirality, if its image in a plane mirror, ideally realized, cannot be brought to coincide with itself.3” As Mislow has pointed out, Kelvin’s geometric definition of chirality is equivalent to that given many years later by Vladimir Prelog in his 1975 Nobel Prize lecture: “An object is chiral if it cannot be brought into congruence with its mirror image by translation or rotation.” Chirality is defined as a property of object which is non superimposable with its mirror image resulting in a pair of stereoisomers referred as enantiomers.