Does Oxygen Feature Chalcogen Bonding?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Dependence of the Sio Bond Length on Structural Parameters in Coesite, the Silica Polymorphs, and the Clathrasils
American Mineralogist, Volume 75, pages 748-754, 1990 The dependence of the SiO bond length on structural parameters in coesite, the silica polymorphs, and the clathrasils M. B. BOISEN, JR. Department of Mathematics, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, U.S.A. G. V. GIBBS, R. T. DOWNS Department of Geological Sciences, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, U.S.A. PHILIPPE D' ARCO Laboratoire de Geologie, Ecole Normale Superieure, 75230 Paris Cedex OS, France ABSTRACT Stepwise multiple regression analyses of the apparent R(SiO) bond lengths were com- pleted for coesite and for the silica polymorphs together with the clathrasils as a function of the variables};(O), P (pressure), f,(Si), B(O), and B(Si). The analysis of 94 bond-length data recorded for coesite at a variety of pressures and temperatures indicates that all five of these variables make a significant contribution to the regression sum of squares with an R2 value of 0.84. A similar analysis of 245 R(SiO) data recorded for silica polymorphs and clathrasils indicate that only three of the variables (B(O), };(O), and P) make a signif- icant contribution to the regression with an R2 value of 0.90. The distribution of B(O) shows a wide range of values from 0.25 to 10.0 A2 with nearly 80% of the observations clustered between 0.25 and 3.0 A2 and the remaining values spread uniformly between 4.5 and 10.0 A2. A regression analysis carried out for these two populations separately indicates, for the data set with B(O) values less than three, that };(O) B(O), P, and };(Si) are all significant with an R2 value of 0.62. -

The Periodic Electronegativity Table
The Periodic Electronegativity Table Jan C. A. Boeyens Unit for Advanced Study, University of Pretoria, South Africa Reprint requests to J. C. A. Boeyens. E-mail: [email protected] Z. Naturforsch. 2008, 63b, 199 – 209; received October 16, 2007 The origins and development of the electronegativity concept as an empirical construct are briefly examined, emphasizing the confusion that exists over the appropriate units in which to express this quantity. It is shown how to relate the most reliable of the empirical scales to the theoretical definition of electronegativity in terms of the quantum potential and ionization radius of the atomic valence state. The theory reflects not only the periodicity of the empirical scales, but also accounts for the related thermochemical data and serves as a basis for the calculation of interatomic interaction within molecules. The intuitive theory that relates electronegativity to the average of ionization energy and electron affinity is elucidated for the first time and used to estimate the electron affinities of those elements for which no experimental measurement is possible. Key words: Valence State, Quantum Potential, Ionization Radius Introduction electronegative elements used to be distinguished tra- ditionally [1]. Electronegativity, apart from being the most useful This theoretical notion, in one form or the other, has theoretical concept that guides the practising chemist, survived into the present, where, as will be shown, it is also the most bothersome to quantify from first prin- provides a precise definition of electronegativity. Elec- ciples. In historical context the concept developed in a tronegativity scales that fail to reflect the periodicity of natural way from the early distinction between antag- the L-M curve will be considered inappropriate. -

Noncovalent Bonds Through Sigma and Pi-Hole Located on the Same Molecule. Guiding Principles and Comparisons
molecules Review Noncovalent Bonds through Sigma and Pi-Hole Located on the Same Molecule. Guiding Principles and Comparisons Wiktor Zierkiewicz 1,* , Mariusz Michalczyk 1,* and Steve Scheiner 2 1 Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeze˙ Wyspia´nskiego27, 50-370 Wrocław, Poland 2 Department of Chemistry and Biochemistry, Utah State University Logan, Logan, UT 84322-0300, USA; [email protected] * Correspondence: [email protected] (W.Z.); [email protected] (M.M.) Abstract: Over the last years, scientific interest in noncovalent interactions based on the presence of electron-depleted regions called σ-holes or π-holes has markedly accelerated. Their high directionality and strength, comparable to hydrogen bonds, has been documented in many fields of modern chemistry. The current review gathers and digests recent results concerning these bonds, with a focus on those systems where both σ and π-holes are present on the same molecule. The underlying principles guiding the bonding in both sorts of interactions are discussed, and the trends that emerge from recent work offer a guide as to how one might design systems that allow multiple noncovalent bonds to occur simultaneously, or that prefer one bond type over another. Keywords: molecular electrostatic potential; halogen bond; pnicogen bond; tetrel bond; chalcogen bond; cooperativity Citation: Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Noncovalent Bonds through Sigma and Pi-Hole Located on the Same 1. Introduction Molecule. Guiding Principles and The concept of the σ-hole, introduced to a wide audience in 2005 at a conference in Comparisons. Molecules 2021, 26, Prague by Tim Clark [1], influenced a way of thinking about noncovalent interactions 1740. -

No Rabbit Ears on Water. the Structure of the Water Molecule
No Rabbit Ears on Water The Structure of the Water Molecule: What Should We Tell the Students? Michael Laing University of Natal. Durban, South Africa In the vast majority of high school (I)and freshman uni- He also considers the possibility of s character in the bond- versity (2) chemistry textbooks, water is presented as a bent ing orbitals of OF2 and OH2 to improve the "strength" of the molecule whose bonding can be described bv the Gilles~ie- orbital (p 111) from 1.732 for purep to 2.00 for the ideal sp3. Nyholm approach with the oxygen atomsp3 Gyhridized. The Inclusion of s character in the hybrid will be "resisted in the lone uairs are said to he dominant causine the H-O-H anele case of atoms with an nnshared pair.. in the s orbital, to beieduced from the ideal 1091120to 104' and are regul&ly which is more stable than the p orbitals" (p 120). Estimates shown as equivalent in shape and size even being described of the H-E-H angle range from 91.2 for AsH3 to 93.5O for as "rabbit ears" (3). Chemists have even been advised to Hz0. He once again ascribes the anomalies for Hz0 to "re- wear Mickey Mouse ears when teaching the structure and uulsion of the charzes of the hvdroaeu atoms" (D 122). bonding of the H20molecule, presumably to emphasize the - In his famous hook (6)~itac~oro&kii describks the bond- shape of the molecule (and possibly the two equivalent lone ing in water and HzS (p 10). -

Removal of Heavy Metals from Aqueous Solution by Zeolite in Competitive Sorption System
International Journal of Environmental Science and Development, Vol. 3, No. 4, August 2012 Removal of Heavy Metals from Aqueous Solution by Zeolite in Competitive Sorption System Sabry M. Shaheen, Aly S. Derbalah, and Farahat S. Moghanm rich volcanic rocks (tuff) with fresh water in playa lakes or Abstract—In this study, the sorption behaviour of natural by seawater [5]. (clinoptilolite) zeolites with respect to cadmium (Cd), copper The structures of zeolites consist of three-dimensional (Cu), nickel (Ni), lead (Pb) and zinc (Zn) has been studied in frameworks of SiO and AlO tetrahedra. The aluminum ion order to consider its application to purity metal finishing 4 4 wastewaters. The batch method has been employed, using is small enough to occupy the position in the center of the competitive sorption system with metal concentrations in tetrahedron of four oxygen atoms, and the isomorphous 4+ 3+ solution ranging from 50 to 300 mg/l. The percentage sorption replacement of Si by Al produces a negative charge in and distribution coefficients (Kd) were determined for the the lattice. The net negative charge is balanced by the sorption system as a function of metal concentration. In exchangeable cation (sodium, potassium, or calcium). These addition lability of the sorbed metals was estimated by DTPA cations are exchangeable with certain cations in solutions extraction following their sorption. The results showed that Freundlich model described satisfactorily sorption of all such as lead, cadmium, zinc, and manganese [6]. The fact metals. Zeolite sorbed around 32, 75, 28, 99, and 59 % of the that zeolite exchangeable ions are relatively innocuous added Cd, Cu, Ni, Pb and Zn metal concentrations (sodium, calcium, and potassium ions) makes them respectively. -
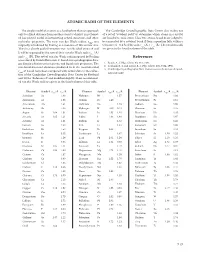
ATOMIC RADII of the ELEMENTS References
ATOMIC RADII OF THE ElEMENTS The simple model of an atom as a hard sphere that can approach The Cambridge Crystallographic Data Center also makes use only to a fixed distance from another atom to which it is not bond- of a set of “covalent radii” to determine which atoms in a crystal ed has proved useful in interpreting crystal structures and other are bonded to each other . Thus two atoms A and B are judged to molecular properties . The term van der Waals radius, rvdw, was be connected by a covalent bond if their separation falls within a originally introduced by Pauling as a measure of this atomic size . tolerance of ±0 .4 Å of the sum rcov (A) + rcov (B) . The covalent radii Thus in a closely packed structure two non-bonded atoms A and are given in the fourth column of the table . B will be separated by the sum of their van der Waals radii rvdw (A) and rvdw (B) . The set of van der Waals radii proposed by Pauling References was refined by Bondi (Reference 1) based on crystallographic data, gas kinetic collision cross sections, and liquid state properties . The 1 . Bondi, A ., J. Phys. Chem. 68, 441, 1964 . non-bonded contact distances predicted from the recommended 2 . Rowland, R . S . and Taylor, R ., J. Phys. Chem. 100, 7384, 1996 . 3 . Cambridge Crystallographic Data Center, www .ccdc .cam .ac .uk/prod- r of Bondi have been compared with actual data in the collec- vdw ucts/csd/radii/ tion of the Cambridge Crystallographic Data Center by Rowland and Taylor (Reference 2) and modified slightly . -

The Nafure of Silicon-Oxygen Bonds
AmericanMineralogist, Volume 65, pages 321-323, 1980 The nafure of silicon-oxygenbonds LtNus PeurrNc Linus Pauling Institute of Scienceand Medicine 2700 Sand Hill Road, Menlo Park, California 94025 Abstract Donnay and Donnay (1978)have statedthat an electron-densitydetermination carried out on low-quartz by R. F. Stewart(personal communication), which assignscharges -0.5 to the oxygen atoms and +1.0 to the silicon atoms, showsthe silicon-oxygenbond to be only 25 percentionic, and hencethat "the 50/50 descriptionofthe past fifty years,which was based on the electronegativity difference between Si and O, is incorrect." This conclusion, however, ignoresthe evidencethat each silicon-oxygenbond has about 55 percentdouble-bond char- acter,the covalenceof silicon being 6.2,with transferof 2.2 valenc,eelectrons from oxygen to silicon. Stewart'svalue of charge*1.0 on silicon then leads to 52 percentionic characterof the bonds, in excellentagreement with the value 5l percent given by the electronegativity scale. Donnay and Donnay (1978)have recently referred each of the four surrounding oxygen atoms,this ob- to an "absolute" electron-densitydetermination car- servationleads to the conclusionthat the amount of ried out on low-quartz by R. F. Stewart, and have ionic character in the silicon-oxygsa 5ingle bond is statedthat "[t showsthe Si-O bond to be 75 percent 25 percent. It is, however,not justified to make this covalent and only 25 percent ionic; the 50/50 de- assumption. scription of the past fifty years,which was basedon In the first edition of my book (1939)I pointed out the electronegativitydifference between Si and O, is that the observedSi-O bond length in silicates,given incorrect." as 1.604, is about 0.20A less than the sum of the I formulated the electronegativityscale in 1932, single-bond radii of the two atoms (p. -

The Hydrogen Bond: a Hundred Years and Counting
J. Indian Inst. Sci. A Multidisciplinary Reviews Journal ISSN: 0970-4140 Coden-JIISAD © Indian Institute of Science 2019. The Hydrogen Bond: A Hundred Years and Counting Steve Scheiner* REVIEW REVIEW ARTICLE Abstract | Since its original inception, a great deal has been learned about the nature, properties, and applications of the H-bond. This review summarizes some of the unexpected paths that inquiry into this phenom- enon has taken researchers. The transfer of the bridging proton from one molecule to another can occur not only in the ground electronic state, but also in various excited states. Study of the latter process has devel- oped insights into the relationships between the nature of the state, the strength of the H-bond, and the height of the transfer barrier. The enormous broadening of the range of atoms that can act as both pro- ton donor and acceptor has led to the concept of the CH O HB, whose ··· properties are of immense importance in biomolecular structure and function. The idea that the central bridging proton can be replaced by any of various electronegative atoms has fostered the rapidly growing exploration of related noncovalent bonds that include halogen, chalco- gen, pnicogen, and tetrel bonds. Keywords: Halogen bond, Chalcogen bond, Pnicogen bond, Tetrel bond, Excited-state proton transfer, CH O H-bond ··· 1 Introduction charge on the proton. The latter can thus attract Over the course of its century of study following the partial negative charge of an approaching its earliest conceptual formulation1,2, the hydro- nucleophile D. Another important factor resides gen bond (HB) has surrendered many of the in the perturbation of the electronic structure mysteries of its source of stability and its myriad of the two species as they approach one another. -

Atomic Structures of the M Olecular Components in DNA and RNA Based on Bond Lengths As Sums of Atomic Radii
1 Atomic St ructures of the M olecular Components in DNA and RNA based on Bond L engths as Sums of Atomic Radii Raji Heyrovská (Institute of Biophysics, Academy of Sciences of the Czech Republic) E-mail: [email protected] Abstract The author’s interpretation in recent years of bond lengths as sums of the relevant atomic/ionic radii has been extended here to the bonds in the skeletal structures of adenine, guanine, thymine, cytosine, uracil, ribose, deoxyribose and phosphoric acid. On examining the bond length data in the literature, it has been found that the averages of the bond lengths are close to the sums of the corresponding atomic covalent radii of carbon (tetravalent), nitrogen (trivalent), oxygen (divalent), hydrogen (monovalent) and phosphorus (pentavalent). Thus, the conventional molecular structures have been resolved here (for the first time) into probable atomic structures. 1. Introduction The interpretation of the bond lengths in the molecules constituting DNA and RNA have been of interest ever since the discovery [1] of the molecular structure of nucleic acids. This work was motivated by the earlier findings [2a,b] that the inter-atomic distance between any two atoms or ions is, in general, the sum of the radii of the atoms (and or ions) constituting the chemical bond, whether the bond is partially or fully ionic or covalent. It 2 was shown in [3] that the lengths of the hydrogen bonds in the Watson- Crick [1] base pairs (A, T and C, G) of nucleic acids as well as in many other inorganic and biochemical compounds are sums of the ionic or covalent radii of the hydrogen donor and acceptor atoms or ions and of the transient proton involved in the hydrogen bonding. -

Chemical Physics 524 (2019) 55–62
Chemical Physics 524 (2019) 55–62 Contents lists available at ScienceDirect Chemical Physics journal homepage: www.elsevier.com/locate/chemphys Structures of clusters surrounding ions stabilized by hydrogen, halogen, T chalcogen, and pnicogen bonds ⁎ ⁎ Steve Scheinera, , Mariusz Michalczykb, Wiktor Zierkiewiczb, a Department of Chemistry and Biochemistry, Utah State University Logan, Utah 84322-0300, United States b Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland ARTICLE INFO ABSTRACT Keywords: Four H-binding HCl and HF molecules position themselves at the vertices of a tetrahedron when surrounding a Tetrahedral central Cl−. Halogen bonding BrF and ClF form a slightly distorted tetrahedron, a tendency that is amplified for QTAIM ClCN which forms a trigonal pyramid. Chalcogen bonding SF2, SeF2, SeFMe, and SeCSe occupy one hemisphere NBO of the central ion, leaving the other hemisphere empty. This pattern is repeated for pnicogen bonding PF3, SeF3 Cl− and AsCF. The clustering of solvent molecules on one side of the Cl− is attributed to weak attractive interactions Na+ between them, including chalcogen, pnicogen, halogen, and hydrogen bonds. Binding energies of four solvent molecules around a central Na+ are considerably reduced relative to chloride, and the geometries are different, with no empty hemisphere. The driving force maximizes the number of electronegative (F or O) atoms close to the Na+, and the presence of noncovalent bonds between solvent molecules. 1. Introduction particular, the S, Se, etc group can engage in chalcogen bonds (YBs) [19–25], and the same is true of the P, As family which forms pnicogen The hydrogen bond (HB) is arguably the most important and in- bonds (ZBs) [26–34]. -

Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc
Metal-Metal (MM) Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc Richard H. Duncan Lyngdoh*,a, Henry F. Schaefer III*,b and R. Bruce King*,b a Department of Chemistry, North-Eastern Hill University, Shillong 793022, India B Centre for Computational Quantum Chemistry, University of Georgia, Athens GA 30602 ABSTRACT: This survey of metal-metal (MM) bond distances in binuclear complexes of the first row 3d-block elements reviews experimental and computational research on a wide range of such systems. The metals surveyed are titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, and zinc, representing the only comprehensive presentation of such results to date. Factors impacting MM bond lengths that are discussed here include (a) n+ the formal MM bond order, (b) size of the metal ion present in the bimetallic core (M2) , (c) the metal oxidation state, (d) effects of ligand basicity, coordination mode and number, and (e) steric effects of bulky ligands. Correlations between experimental and computational findings are examined wherever possible, often yielding good agreement for MM bond lengths. The formal bond order provides a key basis for assessing experimental and computationally derived MM bond lengths. The effects of change in the metal upon MM bond length ranges in binuclear complexes suggest trends for single, double, triple, and quadruple MM bonds which are related to the available information on metal atomic radii. It emerges that while specific factors for a limited range of complexes are found to have their expected impact in many cases, the assessment of the net effect of these factors is challenging. -
![Atomic and Ionic Radii of Elements 1–96 Martinrahm,*[A] Roald Hoffmann,*[A] and N](https://docslib.b-cdn.net/cover/8398/atomic-and-ionic-radii-of-elements-1-96-martinrahm-a-roald-hoffmann-a-and-n-668398.webp)
Atomic and Ionic Radii of Elements 1–96 Martinrahm,*[A] Roald Hoffmann,*[A] and N
DOI:10.1002/chem.201602949 Full Paper & Elemental Radii Atomic and Ionic Radii of Elements 1–96 MartinRahm,*[a] Roald Hoffmann,*[a] and N. W. Ashcroft[b] Abstract: Atomic and cationic radii have been calculated for tive measureofthe sizes of non-interacting atoms, common- the first 96 elements, together with selected anionicradii. ly invoked in the rationalization of chemicalbonding, struc- The metric adopted is the average distance from the nucleus ture, and different properties. Remarkably,the atomic radii where the electron density falls to 0.001 electrons per bohr3, as defined in this way correlate well with van der Waals radii following earlier work by Boyd. Our radii are derived using derived from crystal structures. Arationalizationfor trends relativistic all-electron density functional theory calculations, and exceptionsinthose correlations is provided. close to the basis set limit. They offer asystematic quantita- Introduction cule,[2] but we prefer to follow through with aconsistent pic- ture, one of gauging the density in the atomic groundstate. What is the size of an atom or an ion?This question has been The attractivenessofdefining radii from the electron density anatural one to ask over the centurythat we have had good is that a) the electron density is, at least in principle, an experi- experimental metricinformation on atoms in every form of mental observable,and b) it is the electron density at the out- matter,and (more recently) reliable theory for thesesame ermost regionsofasystem that determines Pauli/exchange/ atoms. And the momentone asks this question one knows same-spinrepulsions, or attractive bondinginteractions, with that there is no unique answer.Anatom or ion coursing down achemical surrounding.