Nocturnal Frontal Lobe Epilepsy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

EEG in the Diagnosis, Classification, and Management of Patients With
EEG IN THE DIAGNOSIS, J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.2005.069245 on 16 June 2005. Downloaded from CLASSIFICATION, AND MANAGEMENT ii2 OF PATIENTS WITH EPILEPSY SJMSmith J Neurol Neurosurg Psychiatry 2005;76(Suppl II):ii2–ii7. doi: 10.1136/jnnp.2005.069245 he human electroencephalogram (EEG) was discovered by the German psychiatrist, Hans Berger, in 1929. Its potential applications in epilepsy rapidly became clear, when Gibbs and Tcolleagues in Boston demonstrated 3 per second spike wave discharge in what was then termed petit mal epilepsy. EEG continues to play a central role in diagnosis and management of patients with seizure disorders—in conjunction with the now remarkable variety of other diagnostic techniques developed over the last 30 or so years—because it is a convenient and relatively inexpensive way to demonstrate the physiological manifestations of abnormal cortical excitability that underlie epilepsy. However, the EEG has a number of limitations. Electrical activity recorded by electrodes placed on the scalp or surface of the brain mostly reflects summation of excitatory and inhibitory postsynaptic potentials in apical dendrites of pyramidal neurons in the more superficial layers of the cortex. Quite large areas of cortex—in the order of a few square centimetres—have to be activated synchronously to generate enough potential for changes to be registered at electrodes placed on the scalp. Propagation of electrical activity along physiological pathways or through volume conduction in extracellular spaces may give a misleading impression as to location of the source of the electrical activity. Cortical generators of the many normal and abnormal cortical activities recorded in the EEG are still largely unknown. -

Dictionary of Epilepsy
DICTIONARY OF EPILEPSY PART I: DEFINITIONS .· DICTIONARY OF EPILEPSY PART I: DEFINITIONS PROFESSOR H. GASTAUT President, University of Aix-Marseilles, France in collaboration with an international group of experts ~ WORLD HEALTH- ORGANIZATION GENEVA 1973 ©World Health Organization 1973 Publications of the World Health Organization enjoy copyright protection in accord ance with the provisions of Protocol 2 of the Universal Copyright Convention. For rights of reproduction or translation of WHO publications, in part or in toto, application should be made to the Office of Publications and Translation, World Health Organization, Geneva, Switzerland. The World Health Organization welcomes such applications. PRINTED IN SWITZERLAND WHO WORKING GROUP ON THE DICTIONARY OF EPILEPSY1 Professor R. J. Broughton, Montreal Neurological Institute, Canada Professor H. Collomb, Neuropsychiatric Clinic, University of Dakar, Senegal Professor H. Gastaut, Dean, Joint Faculty of Medicine and Pharmacy, University of Aix-Marseilles, France Professor G. Glaser, Yale University School of Medicine, New Haven, Conn., USA Professor M. Gozzano, Director, Neuropsychiatric Clinic, Rome, Italy Dr A. M. Lorentz de Haas, Epilepsy Centre "Meer en Bosch", Heemstede, Netherlands Professor P. Juhasz, Rector, University of Medical Science, Debrecen, Hungary Professor A. Jus, Chairman, Psychiatric Department, Academy of Medicine, Warsaw, Poland Professor A. Kreindler, Institute of Neurology, Academy of the People's Republic of Romania, Bucharest, Romania Dr J. Kugler, Department of Psychiatry, University of Munich, Federal Republic of Germany Dr H. Landolt, Medical Director, Swiss Institute for Epileptics, Zurich, Switzerland Dr B. A. Lebedev, Chief, Mental Health, WHO, Geneva, Switzerland Dr R. L. Masland, Department of Neurology, College of Physicians and Surgeons, Columbia University, New York, USA Professor F. -

Nocturnal Epilepsy and Parasomnias Dr.Ram Sankaraneni 10/02/2020
10/2/2020 Nocturnal Epilepsy and Parasomnias Dr.Ram Sankaraneni 10/02/2020 1 CREIGHTON UNIVERSITY Disclosures • None 2 1 10/2/2020 CREIGHTON UNIVERSITY Epilepsy and Sleep • 7.5 to 45 percent of people who have epilepsy have seizures mostly during sleep • Sleep disorders more prevalent in epileptic population 3 CREIGHTON UNIVERSITY • Diagnosis of complex nocturnal behaviors is difficult 4 2 10/2/2020 CREIGHTON UNIVERSITY Differential Diagnosis • Nocturnal Seizures • Non Epileptic Motor Disorders 5 CREIGHTON UNIVERSITY Non Epileptic Motor Disorders • NREM Parasomnias • REM parasomnias • Sleep related movement disorders • Psychogenic 6 3 10/2/2020 CREIGHTON UNIVERSITY When to Suspect an Epileptic etiology • Stereotyped nature • High frequency/Clusters • Timing of the events • Duration • Age of onset 7 CREIGHTON UNIVERSITY Nocturnal Seizures • Clinical manifestation of seizures vary based on location and involved network 8 4 10/2/2020 CREIGHTON UNIVERSITY Seizures in various stages of sleep 70 60 50 40 %seizures 30 %sleep 20 10 0 stage 1 stage 2 SWS REM Herman et al, Neurology 2001;56:1453-9. 9 Percentage of partial seizures arising during sleep from various seizure onset zones. S.T. Herman et al. Neurology 2001;56:1453-1459 ©2001 by Lippincott Williams & Wilkins 10 5 10/2/2020 CREIGHTON UNIVERSITY Frontal Lobe Epilepsy •2nd most common epilepsy ( 18% of adults) • Often misdiagnosed 11 CREIGHTON UNIVERSITY Challenges in Diagnosis • Dramatic/complex movements • No postictal state • Often no Aura • EEG – excessive artifacts • EEG – can be normal -

Nocturnal Epilepsies in Adults
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Seizure 1997; 6: 145-149 Nocturnal epilepsies in adults BASIM A YAQUB, GHAZALA WAHEED & MOHAMMAD MU KABIRAJ Division of Neurology and Neurophysiology, King Khalid University Hospital, PO Box 7805, Riyadh 11472, Saudi Arabia Correspondence to: Dr. Basim A. Yaqub, Consultant Neurologist, Department of Clinical Neurosciences. Armed Forces Hospital, P.O. Box 7897 (X1006), Riyadh 11159, Kingdom of Saudi Arabia We evaluated the clinical characteristics and the electroencephalographic (EEG) findings by long video-EEG monitoring in 64 successive patients with definite nocturnal seizures. Mental state, neurological examination, neuroimaging and EEG background were normal in all patients. Classification of epilepsies was possible in 42 out of 64 (66%) patients according to the revised Classification of Epilepsies and Epileptic Syndromes by the Commission on Classification and Terminology of International League Against Epilepsy (1989). Out of those 42 patients, 33 (79%) had partial epilepsies, while 9 (21%) had generalized epilepsies. Response to antiepileptic drugs was excellent and only 4 (6%) patients had one seizure attack per year, two of them were on two antiepileptic drugs while the others were free of seizure on a single drug during the 2 years of follow-up. It seems that nocturnal seizuresin adults form a new distinctive partial epileptic syndrome of a benign entity. Key words: nocturnal seizures: epileptic syndromes: video-EEG monitoring: benign childhood epilepsy with centro-temporal spikes:antiepileptic drugs. INTRODUCTION EEG (VEEG) monitoring, has become an essential technique in most sleep laboratories, The pattern of seizure occurrence in most also it is an important technique for the epileptics is random without cycling or clustering identification and classification of unusual sei- so they can occur during the daytime or sleep’. -
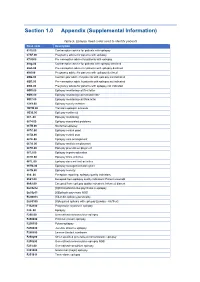
Section 1.0 Appendix (Supplemental Information)
Section 1.0 Appendix (Supplemental Information) Table 5. Epilepsy Read codes used to identify patients Read code Description 6110.00 Contraceptive advice for patients with epilepsy 67AF.00 Pregnancy advice for patients with epilepsy 67IJ000 Pre-conception advice for patients with epilepsy 8IAg.00 Contraceptive advice for patients with epilepsy declined 8IAh.00 Pre-conception advice for patients with epilepsy declined 8IAi.00 Pregnancy advice for patients with epilepsy declined 8IB2.00 Contraceptiv advice for patients with epilepsy not indicated 8IB3.00 Pre-conception advic fr patients with epilepsy not indicated 8IB4.00 Pregnancy advice for patients with epilepsy not indicated 9Of5.00 Epilepsy monitoring call first letter 9Of6.00 Epilepsy monitoring call second letter 9Of7.00 Epilepsy monitoring call third letter 13Y9.00 Epilepsy society member 1B1W.00 Transient epileptic amnesia 1O30.00 Epilepsy confirmed 667..00 Epilepsy monitoring 6674.00 Epilepsy associated problems 667B.00 Nocturnal epilepsy 667C.00 Epilepsy control good 667D.00 Epilepsy control poor 667E.00 Epilepsy care arrangement 667G.00 Epilepsy restricts employment 667H.00 Epilepsy prevents employment 667J.00 Epilepsy impairs education 667K.00 Epilepsy limits activities 667L.00 Epilepsy does not limit activities 667M.00 Epilepsy management plan given 667N.00 Epilepsy severity 9h6..00 Exception reporting: epilepsy quality indicators 9h61.00 Excepted from epilepsy quality indicators: Patient unsuitabl 9h62.00 Excepted from epilepsy quality indicators: Informed dissent Eu05212 -

Reduction of Rapid Eye Movement Sleep by Diurnal and Nocturnal Seizures in Temporal Lobe Epilepsy
ORIGINAL CONTRIBUTION Reduction of Rapid Eye Movement Sleep by Diurnal and Nocturnal Seizures in Temporal Lobe Epilepsy Carl W. Bazil, MD, PhD; Luiz H. M. Castro, MD; Thaddeus S. Walczak, MD Background: Patients with brief, complex partial sei- maintenance of wakefulness test and 2 subjective drowsi- zures frequently suffer from tiredness and decreased pro- ness tests. ductivity that continue well beyond the postictal pe- riod. A possible explanation is that seizures, even when Results: Daytime seizures reduced REM from 18% ± 1% occurring during the day, disrupt sleep the following night. to 12% ± 2% (P = .003). Night seizures reduced REM from 16%±1%to6.8%±2%(P,.001). Night seizures also sig- Objective: To determine the effect of temporal lobe com- nificantly reduced stages 2 and 4 while increasing stage plex partial seizures on sleep structure and daytime 1 sleep. Night seizures, but not day seizures, signifi- drowsiness. cantly reduced sleep efficiency, increased time to first REM period, and increased drowsiness as measured by the Methods: Patients with temporal lobe epilepsy were ad- maintenance of wakefulness test. mitted for video-electroencephalography monitoring. All- night polysomnography was recorded under the follow- Conclusions: Temporal lobe complex partial seizures de- ing 3 conditions: seizure free, seizure during the day before crease REM sleep, particularly when occurring during sleep the recording, and seizure during the recording. Percent- but also when occurring on the previous day. This may, age of time in each sleep stage, sleep efficiency, and time in part, be responsible for the prolonged impairment of func- to first and second rapid eye movement (REM) period tioning that some patients report following seizures. -

Epilepsy 2017 from Bench to Bedside 23–24 September 2017 Eaching Weekend, Weekend, Eaching
EPILEPSY 2017 FROM BENCH TO BEDSIDE 23–24 SEPTEMBER 2017 EACHING WEEKEND, WEEKEND, EACHING A PRACTICAL GUIDE TO EPILEPSY ILAE SPR T Edited by XVI F.J. Rugg-Gunn and H.B. Stapley International League Against Epilepsy EPILEPSY 2017 From Bench to Bedside A Practical Guide to Epilepsy Lecture Notes Sixteenth Epilepsy Teaching Weekend 23–24 September 2017 University of Oxford Mathematical Institute Edited by F.J. Rugg-Gunn and H.B. Stapley PREVIOUS PREVIOUS NEXT NEXT < SECTION < CHAPTER CONTENTS CHAPTER> SECTION> LECTURE NOTES for the Contents Sixteenth Epilepsy Teaching Weekend 23−24 September 2017 University of Oxford Mathematical Institute Preface F.J. RUGG-GUNN .................................................................................................................................... vi Introduction F.J. RUGG-GUNN ................................................................................................................................... vii Sixteenth Edition (2017) Edited by F.J. Rugg-Gunn and H.B. Stapley Section One – Introduction 1 The incidence and prevalence of epilepsy A. NELIGAN and J.W. SANDER ....................................................................................................3 2 Classification and terminology to organise seizures and epilepsy T. WEHNER ...................................................................................................................................11 Section Two – Basic science 3 Basic mechanisms of epilepsy J.G.R. JEFFERYS ���������������������������������������������������������������������������������������������������������������������������21 -

Pearls & Oy-Sters
RESIDENT & FELLOW SECTION Pearls & Oy-sters: Section Editor Diagnostic challenges in nocturnal frontal John J. Millichap, MD lobe epilepsy Fieke M.E. Cox, MD, PEARLS disorientation. He did not call the nursing staff or PhD • Nocturnal frontal lobe epilepsy (NFLE) is best remember the episodes afterwards. The EEG Gert Jan Lammers, MD, diagnosed by combined EEG-video recording. showed a K-complex at the beginning of each of PhD – • Clinical features of events can contribute to dif- these events, followed by a diffuse 11 12 Hz Roland D. Thijs, MD, ferentiation of NFLE and a parasomnia. rhythm for several seconds, and was once followed PhD by a short delta rhythm over the left frontotemporal Gerhard H. Visser, MD, region (figure). The episodes were accompanied by an OY-STERS PhD acceleration of heart rate (from 54 to 72 bpm). After • In many cases, the scalp EEG is unable to detect the event, when he closed his eyes again, a normal interictal and even ictal abnormalities, because the posterior dominant a rhythm was seen. As the events Correspondence to did not clinically resemble a physiologic arousal (due Dr. Cox: frontal lobe focus may be too deep to be detected. [email protected] to the abrupt onset, marked stereotypy, and the tonic extension of the right hand), and in combination with CASE REPORT A 43-year-old man of normal intelligence consulted our outpatient clinic for the consistent (ictal) EEG findings, these episodes nocturnal events that had started at age 32 years. were considered to be of epileptic origin, and a These arise from sleep when he suddenly awakens, diagnosis of NFLE was made (with paroxysmal experiencing fear and the sensation of falling into a arousals, sometimes with secondary generalization). -
A GUIDE for PARENTS Epilepsyepilepsy
206353_Guide For Parents_Parents 1/14/11 10:47 AM Page 1 A GUIDE FOR PARENTS EpilepsyEpilepsy EPILEPSY EDUCATION SERIES 206353_Guide For Parents_Parents 1/14/11 10:48 AM Page 2 This publication was produced by the The Epilepsy Association of Northern Alberta Phone: 780-488-9600 Toll Free: 1-866-374-5377 Fax: 780-447-5486 Email: [email protected] Website: www.edmontonepilepsy.org This booklet is designed to provide general information about Epilepsy to the public. It does not include specific medical advice, and people with Epilepsy should not make changes based on this information to previously prescribed treatment or activities without first consulting their physician. Special thanks to our Consulting Team, which was comprised of Epilepsy Specialist Neurologists & Neuroscience Nurses, Hospital Epilepsy Clinic Staff, Educators, Individuals with Epilepsy, and Family Members of Individuals with Epilepsy. Free Canada-wide distribution of this publication was made possible by an unrestricted Grant from UCB Canada Inc. © Edmonton Epilepsy Association, 2011 206353_Guide For Parents_Parents 1/14/11 10:48 AM Page 3 Index What is Epilepsy? ______________________________________1 What are the Signs of Childhood Seizures? ________________2 What Causes Epilepsy and Seizures? ______________________3 What are the Different Types of Seizures? __________________5 What are Epilepsies and Epilepsy Syndromes? ____________11 How is Epilepsy Diagnosed? ____________________________17 What is the Treatment for Epilepsy? ______________________23 How Can -

Nocturnal Epilepsy: a Seizure Issue
Editorial Epilepsy Journal Volume 7:3, 2021 ISSN: 2472-0895 Open Access Nocturnal Epilepsy: A Seizure Issue Mohamad Sawan* Professor, Polytechnique Montreal, Canada Nocturnal epilepsy is a seizure issue in which seizures happen while resting, in individuals who experience the ill effects of nocturnal epilepsy, the inside an hour of waking or an hour prior to sleep time. A few basic types of medications are appeared to upset an individual's resting structure. This may epilepsy, including front facing flap epilepsy, can show in a nocturnal state. cause worry in individuals who experience the ill effects of nocturnal epilepsy Epilepsy can be nocturnal if the type of epilepsy triggers seizures just while in light of the fact that undisrupted rest is significant for these individuals, one is snoozing, or on the off chance that one typically has seizures that as it brings down the likeliness of epileptic manifestations to emerge. One happen around then. In the last model, if the subject stays conscious when specific investigation by V. Bradley and D. O'Neill examined the various he is regularly dozing, the subject may have the seizure while alert. Taking types of epilepsy, including nocturnal epilepsy and its relationship with note of this, it is significant for the subject to keep a legitimate resting cycle. rest. They tracked down that a few patients just experienced epileptic side Redirecting from appropriate rest examples can trigger more continuous effects while they are snoozing (nocturnal epilepsy), and that keeping up epileptic side effects in individuals who are determined to have nocturnal great rest helped in decreasing epileptic indications. -

SUPPLEMENTARY INFORMATION Trends in Antiepileptic Drug
Supplementary material BMJ Open SUPPLEMENTARY INFORMATION Trends in antiepileptic drug treatment and effectiveness in clinical practice in England from 2003 to 2016: a retrospective cohort study using electronic medical records Graham Powell, John Logan, Victor Kiri, Simon Borghs BMJ Open https://doi.org/bmjopen-2019-032551 Powell G, et al. BMJ Open 2019; 9:e032551. doi: 10.1136/bmjopen-2019-032551 Supplementary material BMJ Open Supplemental Tables Supplementary Table S1 Diagnostic codes for epilepsy CODES TERM EPILEPSY TYPE International Classification of Diseases, Tenth Revision (ICD-10) G40 Epilepsy Unspecified G40.0 Localisation-related (focal)(partial) idiopathic epilepsy and Partial epileptic syndromes with seizures of localised onset G40.1 Localisation-related (focal)(partial) symptomatic epilepsy and Partial epileptic syndromes with simple partial seizures G40.2 Localisation-related (focal)(partial) symptomatic epilepsy and Partial epileptic syndromes with complex partial seizures G40.3 Generalised idiopathic epilepsy and epileptic syndromes Generalised G40.4 Other generalised epilepsy and epileptic syndromes Generalised G40.5 Special epileptic syndromes Unspecified G40.6 Grand mal seizures, unspecified (with or without petit mal) Generalised G40.7 Petit mal, unspecified, without grand mal seizures Unspecified G40.8 Other epilepsy Unspecified G40.9 Epilepsy, unspecified Unspecified READ codes 1O30.00 Epilepsy confirmed unspecified 667B.00 Nocturnal epilepsy unspecified Eu80300 [X]Acquired aphasia with epilepsy [Landau - Kleffner] -
Redalyc.The Sleep-Wakefulness Cycle of Wistar Rats with Spontaneous Absence-Like Epilepsy
Acta Scientiarum. Biological Sciences ISSN: 1679-9283 [email protected] Universidade Estadual de Maringá Brasil Sanfelice André, Edison; Bruno-Neto, Rafael; do Valle, Angela Cristina; Timo-Iaria, César The sleep-wakefulness cycle of Wistar rats with spontaneous absence-like epilepsy Acta Scientiarum. Biological Sciences, vol. 37, núm. 3, julio-septiembre, 2015, pp. 367- 376 Universidade Estadual de Maringá Maringá, Brasil Available in: http://www.redalyc.org/articulo.oa?id=187142726013 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative Acta Scientiarum http://www.uem.br/acta ISSN printed: 1679-9283 ISSN on-line: 1807-863X Doi: 10.4025/actascibiolsci.v37i3.26908 The sleep-wakefulness cycle of Wistar rats with spontaneous absence-like epilepsy Edison Sanfelice André1*, Rafael Bruno-Neto2, Angela Cristina do Valle3 and César Timo-Iaria3† 1Departamento de Fisioterapia, Universidade Regional de Blumenau, Rua Antônio da Veiga, 140, 89010-971, Blumenau, Santa Catarina, Brazil. 2Departamento de Ciências Morfofisiológicas, Universidade Estadual de Maringá, Maringá, Paraná, Brazil. 3Laboratório de Neurocirurgia Funcional, Divisão de Neurocirurgia, Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil. †In memoriam. *Author for correspondence. E-mail: [email protected] ABSTRACT. Possible interactions between the sleep-wakefulness cycle and a new kind of spontaneous epilepsy, expressed as absence-like seizures and spike-wave bursts in FMUSP rats, are evaluated. The electro-oscillograms of some cortical and subcortical regions of the brain were recorded, as well as head, rostrum/vibrissae and eye movements.