Biotools: Tools Based on Biostrings (Alignment, Classification, Database)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Rare and Abundant Prokaryotic
Diversity of rare and abundant prokaryotic phylotypes in the Prony hydrothermal field and comparison with other serpentinite-hosted ecosystems Eléonore Frouin, Méline Bes, Bernard Ollivier, Marianne Quéméneur, Anne Postec, Didier Debroas, Fabrice Armougom, Gaël Erauso To cite this version: Eléonore Frouin, Méline Bes, Bernard Ollivier, Marianne Quéméneur, Anne Postec, et al.. Diver- sity of rare and abundant prokaryotic phylotypes in the Prony hydrothermal field and compari- son with other serpentinite-hosted ecosystems. Frontiers in Microbiology, Frontiers Media, 2018, 9, 10.3389/fmicb.2018.00102. hal-01734508 HAL Id: hal-01734508 https://hal.archives-ouvertes.fr/hal-01734508 Submitted on 12 Oct 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License fmicb-09-00102 February 3, 2018 Time: 13:27 # 1 ORIGINAL RESEARCH published: 06 February 2018 doi: 10.3389/fmicb.2018.00102 Diversity of Rare and Abundant Prokaryotic Phylotypes in the Prony Hydrothermal Field and Comparison with Other Serpentinite-Hosted Ecosystems Eléonore Frouin1, Méline Bes1, Bernard Ollivier1, Marianne Quéméneur1, Anne Postec1, Didier Debroas2, Fabrice Armougom1 and Gaël Erauso1* 1 Aix-Marseille Univ, Université de Toulon, CNRS, IRD, MIO UM 110, Marseille, France, 2 CNRS UMR 6023, Laboratoire “Microorganismes – Génome et Environnement”, Université Clermont Auvergne, Clermont-Ferrand, France The Bay of Prony, South of New Caledonia, represents a unique serpentinite- hosted hydrothermal field due to its coastal situation. -

Microbial Community Structure Dynamics in Ohio River Sediments During Reductive Dechlorination of Pcbs
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2008 MICROBIAL COMMUNITY STRUCTURE DYNAMICS IN OHIO RIVER SEDIMENTS DURING REDUCTIVE DECHLORINATION OF PCBS Andres Enrique Nunez University of Kentucky Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Nunez, Andres Enrique, "MICROBIAL COMMUNITY STRUCTURE DYNAMICS IN OHIO RIVER SEDIMENTS DURING REDUCTIVE DECHLORINATION OF PCBS" (2008). University of Kentucky Doctoral Dissertations. 679. https://uknowledge.uky.edu/gradschool_diss/679 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Andres Enrique Nunez The Graduate School University of Kentucky 2008 MICROBIAL COMMUNITY STRUCTURE DYNAMICS IN OHIO RIVER SEDIMENTS DURING REDUCTIVE DECHLORINATION OF PCBS ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Agriculture at the University of Kentucky By Andres Enrique Nunez Director: Dr. Elisa M. D’Angelo Lexington, KY 2008 Copyright © Andres Enrique Nunez 2008 ABSTRACT OF DISSERTATION MICROBIAL COMMUNITY STRUCTURE DYNAMICS IN OHIO RIVER SEDIMENTS DURING REDUCTIVE DECHLORINATION OF PCBS The entire stretch of the Ohio River is under fish consumption advisories due to contamination with polychlorinated biphenyls (PCBs). In this study, natural attenuation and biostimulation of PCBs and microbial communities responsible for PCB transformations were investigated in Ohio River sediments. Natural attenuation of PCBs was negligible in sediments, which was likely attributed to low temperature conditions during most of the year, as well as low amounts of available nitrogen, phosphorus, and organic carbon. -

Online Supplementary Figures of Chapter 3
Online Supplementary Figures of Chapter 3 Fabio Gori Figures 1-30 contain pie charts showing the population characterization re- sulting from the taxonomic assignment computed by the methods. On the simulated datasets the true population distribution is also shown. 1 MTR Bacillales (47.11%) Thermoanaerobacterales (0.76%) Clostridiales (33.58%) Lactobacillales (7.99%) Others (10.55%) LCA Bacillales (48.38%) Thermoanaerobacterales (0.57%) Clostridiales (32.14%) Lactobacillales (10.07%) Others (8.84%) True Distribution 333 386 Prochlorales (5.84%) 535 Bacillales (34.61%) Halanaerobiales (4.37%) Thermoanaerobacterales (10.29%) Clostridiales (28.75%) Lactobacillales (9.38%) 1974 Herpetosiphonales (6.77%) 1640 249 587 Figure 1: Population distributions (rank Order) of M1, coverage 0.1x, by MTR and LCA, and the true population distribution. 2 MTR Bacillus (47.34%) Clostridium (14.61%) Lactobacillus (8.71%) Anaerocellum (11.41%) Alkaliphilus (5.14%) Others (12.79%) LCA Bacillus (51.41%) Clostridium (8.08%) Lactobacillus (9.23%) Anaerocellum (15.79%) Alkaliphilus (5.17%) Others (10.31%) True Distribution 386 552 Herpetosiphon (6.77%) 333 Prochlorococcus (5.84%) 587 Bacillus (34.61%) Clostridium (19.07%) Lactobacillus (9.38%) 249 Halothermothrix (4.37%) Caldicellulosiruptor (10.29%) Alkaliphilus (9.68%) 535 1974 1088 Figure 2: Population distributions (rank Genus) of M1, coverage 0.1x, by MTR and LCA, and the true population distribution. 3 MTR Prochlorales (0.07%) Bacillales (47.97%) Thermoanaerobacterales (0.66%) Clostridiales (32.18%) Lactobacillales (7.76%) Others (11.35%) LCA Prochlorales (0.10%) Bacillales (49.02%) Thermoanaerobacterales (0.59%) Clostridiales (30.62%) Lactobacillales (9.50%) Others (10.16%) True Distribution 3293 3950 Prochlorales (5.65%) 5263 Bacillales (36.68%) Halanaerobiales (3.98%) Thermoanaerobacterales (10.56%) Clostridiales (27.34%) Lactobacillales (9.03%) 21382 Herpetosiphonales (6.78%) 15936 2320 6154 Figure 3: Population distributions (rank Order) of M1, coverage 1x, by MTR and LCA, and the true population distribution. -

Metagenomic and PCR-Based Diversity Surveys of [Fefe
ORIGINAL RESEARCH published: 30 August 2016 doi: 10.3389/fmicb.2016.01301 Metagenomic and PCR-Based Diversity Surveys of [FeFe]-Hydrogenases Combined with Isolation of Alkaliphilic Hydrogen-Producing Bacteria from the Serpentinite-Hosted Prony Hydrothermal Field, New Caledonia Nan Mei 1, Anne Postec 1, Christophe Monnin 2, Bernard Pelletier 3, Claude E. Payri 3, Bénédicte Ménez 4, Eléonore Frouin 1, Bernard Ollivier 1, Gaël Erauso 1 and Marianne Quéméneur 1* 1 2 Edited by: Aix Marseille Univ, Université de Toulon, CNRS, IRD, MIO, Marseille, France, GET UMR5563 (Centre National de la 3 Andreas Teske, Recherche Scientifique/UPS/IRD/CNES), Géosciences Environnement Toulouse, Toulouse, France, Institut pour la 4 University of North Carolina at Chapel Recherche et le Développement (IRD) Centre de Nouméa, MIO UM 110, Nouméa, Nouvelle-Calédonie, Institut de Physique Hill, USA du Globe de Paris, Sorbonne Paris Cité, Univ Paris Diderot, Centre National de la Recherche Scientifique, Paris, France Reviewed by: John R. Spear, High amounts of hydrogen are emitted in the serpentinite-hosted hydrothermal field Colorado School of Mines, USA of the Prony Bay (PHF, New Caledonia), where high-pH (∼11), low-temperature Igor Tiago, ◦ University of Coimbra, Portugal (<40 C), and low-salinity fluids are discharged in both intertidal and shallow submarine *Correspondence: environments. In this study, we investigated the diversity and distribution of potentially Marianne Quéméneur hydrogen-producing bacteria in Prony hyperalkaline springs by using metagenomic [email protected] analyses and different PCR-amplified DNA sequencing methods. The retrieved Specialty section: sequences of hydA genes, encoding the catalytic subunit of [FeFe]-hydrogenases and, This article was submitted to used as a molecular marker of hydrogen-producing bacteria, were mainly related to those Extreme Microbiology, a section of the journal of Firmicutes and clustered into two distinct groups depending on sampling locations. -

Global Metagenomic Survey Reveals a New Bacterial Candidate Phylum in Geothermal Springs
ARTICLE Received 13 Aug 2015 | Accepted 7 Dec 2015 | Published 27 Jan 2016 DOI: 10.1038/ncomms10476 OPEN Global metagenomic survey reveals a new bacterial candidate phylum in geothermal springs Emiley A. Eloe-Fadrosh1, David Paez-Espino1, Jessica Jarett1, Peter F. Dunfield2, Brian P. Hedlund3, Anne E. Dekas4, Stephen E. Grasby5, Allyson L. Brady6, Hailiang Dong7, Brandon R. Briggs8, Wen-Jun Li9, Danielle Goudeau1, Rex Malmstrom1, Amrita Pati1, Jennifer Pett-Ridge4, Edward M. Rubin1,10, Tanja Woyke1, Nikos C. Kyrpides1 & Natalia N. Ivanova1 Analysis of the increasing wealth of metagenomic data collected from diverse environments can lead to the discovery of novel branches on the tree of life. Here we analyse 5.2 Tb of metagenomic data collected globally to discover a novel bacterial phylum (‘Candidatus Kryptonia’) found exclusively in high-temperature pH-neutral geothermal springs. This lineage had remained hidden as a taxonomic ‘blind spot’ because of mismatches in the primers commonly used for ribosomal gene surveys. Genome reconstruction from metagenomic data combined with single-cell genomics results in several high-quality genomes representing four genera from the new phylum. Metabolic reconstruction indicates a heterotrophic lifestyle with conspicuous nutritional deficiencies, suggesting the need for metabolic complementarity with other microbes. Co-occurrence patterns identifies a number of putative partners, including an uncultured Armatimonadetes lineage. The discovery of Kryptonia within previously studied geothermal springs underscores the importance of globally sampled metagenomic data in detection of microbial novelty, and highlights the extraordinary diversity of microbial life still awaiting discovery. 1 Department of Energy Joint Genome Institute, Walnut Creek, California 94598, USA. 2 Department of Biological Sciences, University of Calgary, Calgary, Alberta T2N 1N4, Canada. -

EXPERIMENTAL STUDIES on FERMENTATIVE FIRMICUTES from ANOXIC ENVIRONMENTS: ISOLATION, EVOLUTION, and THEIR GEOCHEMICAL IMPACTS By
EXPERIMENTAL STUDIES ON FERMENTATIVE FIRMICUTES FROM ANOXIC ENVIRONMENTS: ISOLATION, EVOLUTION, AND THEIR GEOCHEMICAL IMPACTS By JESSICA KEE EUN CHOI A dissertation submitted to the School of Graduate Studies Rutgers, The State University of New Jersey In partial fulfillment of the requirements For the degree of Doctor of Philosophy Graduate Program in Microbial Biology Written under the direction of Nathan Yee And approved by _______________________________________________________ _______________________________________________________ _______________________________________________________ _______________________________________________________ New Brunswick, New Jersey October 2017 ABSTRACT OF THE DISSERTATION Experimental studies on fermentative Firmicutes from anoxic environments: isolation, evolution and their geochemical impacts by JESSICA KEE EUN CHOI Dissertation director: Nathan Yee Fermentative microorganisms from the bacterial phylum Firmicutes are quite ubiquitous in subsurface environments and play an important biogeochemical role. For instance, fermenters have the ability to take complex molecules and break them into simpler compounds that serve as growth substrates for other organisms. The research presented here focuses on two groups of fermentative Firmicutes, one from the genus Clostridium and the other from the class Negativicutes. Clostridium species are well-known fermenters. Laboratory studies done so far have also displayed the capability to reduce Fe(III), yet the mechanism of this activity has not been investigated -

Biochemical Characterization of Β-Xylan Acting Glycoside
BIOCHEMICAL CHARACTERIZATION OF β-XYLAN ACTING GLYCOSIDE HYDROLASES FROM THE THERMOPHILIC BACTERIUM Caldicellulosiruptor saccharolyticus Thesis Submitted to The School of Engineering of the UNIVERSITY OF DAYTON In Partial Fulfillment of the Requirements for The Degree of Master of Science in Chemical Engineering By Jin Cao Dayton, Ohio December, 2012 BIOCHEMICAL CHARACTERIZATION OF β-XYLAN ACTING GLYCOSIDE HYDROLASES FROM THE THERMOPHILIC BACTERIUM Caldicellulosiruptor saccharolyticus Name: Cao, Jin APPROVED BY: ______________________ ________________________ Donald A. Comfort, Ph.D. Amy Ciric, Ph.D. Advisory Committee Chairman Committee Member Research Advisor & Assistant Professor Lecturer Department of Department of Chemical & Materials Engineering Chemical & Materials Engineering ________________________ Karolyn Hansen, Ph.D. Committee Member Assistant Professor Department of Biology _______________________ ________________________ John G. Weber, Ph.D. Tony E. Saliba, Ph.D. Associate Dean Dean, School of Engineering School of Engineering & Wilke Distinguished Professor ii ABSTRACT BIOCHEMICAL CHARACTERIZATION OF Β-XYLAN ACTING GLYCOSIDE HYDROLASES FROM THE THERMOPHILIC BACTERIUM Caldicellulosiruptor saccharolyticus Name: Cao, Jin University of Dayton Research Advisor: Donald A. Comfort Fossil fuels have been the dominant source for energy around the world since the industrial revolution, however, with the increasing demand for energy and decreasing fossil fuel reserves alternative energy sources must be exploited. Bio-ethanol is a promising prospect for an alternative energy source to petroleum, especially when plant biomass is used as the replacement carbon source. This gives greater benefit for implementation of bioethanol as a viable alternative transport fuel than food stocks such as starch from corn. The implementation of this next generation of bioethanol requires more robust and environmentally friendly methods for degrading cellulose and hemicellulose, the two major components of plant biomass. -

Phylogenomic Analysis Supports the Ancestral Presence of LPS-Outer Membranes in the Firmicutes
Phylogenomic analysis supports the ancestral presence of LPS-outer membranes in the Firmicutes. Luisa Cs Antunes, Daniel Poppleton, Andreas Klingl, Alexis Criscuolo, Bruno Dupuy, Céline Brochier-Armanet, Christophe Beloin, Simonetta Gribaldo To cite this version: Luisa Cs Antunes, Daniel Poppleton, Andreas Klingl, Alexis Criscuolo, Bruno Dupuy, et al.. Phy- logenomic analysis supports the ancestral presence of LPS-outer membranes in the Firmicutes.. eLife, eLife Sciences Publication, 2016, 5, pp.e14589. 10.7554/eLife.14589.020. pasteur-01362343 HAL Id: pasteur-01362343 https://hal-pasteur.archives-ouvertes.fr/pasteur-01362343 Submitted on 8 Sep 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License RESEARCH ARTICLE Phylogenomic analysis supports the ancestral presence of LPS-outer membranes in the Firmicutes Luisa CS Antunes1†, Daniel Poppleton1†, Andreas Klingl2, Alexis Criscuolo3, Bruno Dupuy4, Ce´ line Brochier-Armanet5, Christophe Beloin6, Simonetta Gribaldo1* 1Unite´ de -
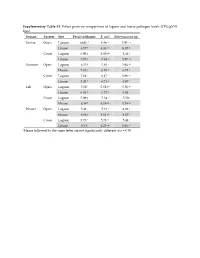
Supplementary Table S1. Fisher Pairwise Comparisons of Lagoon and House Pathogen Levels (CFU/Gvss Log10) Season System Site Fecal Coliforms E
Supplementary Table S1. Fisher pairwise comparisons of lagoon and house pathogen levels (CFU/gVSS log10) Season System Site Fecal coliforms E. coli Enterococcus sp. Spring Open Lagoon 6.00 g1 5.46 hi 5.91 cd House 6.57 e 6.33 cd 6.35 b Cover Lagoon 6.00 g 5.60 gh 5.44 f House 7.73 a 7.63 a 5.87 cd Summer Open Lagoon 6.35 f 5.83 f 5.94 cd House 7.83 a 6.10 e 6.73 a Cover Lagoon 7.04 c 6.47 c 5.86 cd House 7.42 b 6.71 b 6.07 c Fall Open Lagoon 5.58 i 5.58 gh 5.56 ef House 6.81 d 6.72 b 6.01 c Cover Lagoon 5.99 g 5.34 i 5.50 f House 6.38 f 6.19 de 5.74 de Winter Open Lagoon 5.41 j 5.13 j 4.88 g House 6.06 g 5.61 gh 6.67 a Cover Lagoon 5.73 h 5.73 fg 5.44 f House 6.34 f 6.25 de 5.86 cd 1Means followed by the same letter are not significantly different at p = 0.05 Supplementary Table S2. Relative abundances of OTUs identified using universal bacterial primer set, presented as relative abundances (%) Only bacterial families are counted in Figure 5 and discussion involving bacterial family identification. # Identified in # Identified from all 8 All 16 All #OTU Table SpOLRA SuOLRA FOLRA WOLRA SpCLRA SuCLRA FCLRA WCLRA SpOHRA SuOHRA FOHRA WOHRA SpCHRA SuCHRA FCHRA WCHRA all samples lagoon samples Samples Lagoons Notes Key Patulibacteraceae 0 9.93739E‐05* 0.000134 0.000116 0 0 0 0 0 0 0.000452 0 0 0 0 0 4 3 Sp = Spring O = Open Ruaniaceae 0 0 0 0 0 0 0 0 0 0 0 0 0 0.000221577 0 7.73E‐05 2 0 Su = Summer C = Covered Thermoactinomycetaceae 0 0 0.000267 0.000116 0 0.00037092 0.000529 0.000526 0 0.000342922 0.000271 0.000197 0 0 0 0 8 5 F = Fall L = Lagoon Beutenbergiaceae 0.000226334 0.000331246 0.000579 0.001663 0.000620176 0 0.000106 0.000807 0.002320743 0.000571537 0.000723 0.000591 0.000169731 0 0.000606 0 13 7 W = Winter H = House Thermoanaerobacterales Family III. -

Composition of Rabbit Caecal Microbiota and the Effects of Dietary Quercetin Supplementation and Sex Thereupon North M.K
W orld World Rabbit Sci. 2019, 27: 185-198 R abbit doi:10.4995/wrs.2019.11905 Science © WRSA, UPV, 2003 COMPOSITION OF RABBIT CAECAL MICROBIOTA AND THE EFFECTS OF DIETARY QUERCETIN SUPPLEMENTATION AND SEX THEREUPON NORTH M.K. *, DALLE ZOTTE A. †, HOFFMAN L.C. *‡ *Department of Animal Sciences, Stellenbosch University, Private Bag X1, Matieland, STELLENBOSCH 7602, South Africa. †Department of Animal Medicine, Production and Health, University of Padova, Agripolis, Viale dell’Università, 16, 35020 LEGNARO, Padova, Italy. ‡Centre for Nutrition and Food Sciences, Queensland Alliance for Agriculture and Food Innovation (QAAFI), The University of Queensland, Health and Food Sciences Precinct, 39 Kessels Road, COOPERS PLAINS 4108, Australia. Abstract: The purpose of this study was to add to the current understanding of rabbit caecal microbiota. This involved describing its microbial composition and linking this to live performance parameters, as well as determining the effects of dietary quercetin (Qrc) supplementation (2 g/kg feed) and sex on the microbial population. The weight gain and feed conversion ratio of twelve New Zealand White rabbits was measured from 5 to 12 wk old, blood was sampled at 11 wk old for the determination of serum hormone levels, and the rabbits were slaughtered and caecal samples collected at 13 wk old. Ion 16STM metagenome sequencing was used to determine the microbiota profile. The dominance of Firmicutes (72.01±1.14% of mapped reads), Lachnospiraceae (23.94±1.01%) and Ruminococcaceae (19.71±1.07%) concurred with previous reports, but variation both between studies and individual rabbits was apparent beyond this. Significant correlations between microbial families and live performance parameters were found, suggesting that further research into the mechanisms of these associations could be useful. -

Microbial Ecology of Hydraulic Fracturing: Implications for Sustainable Resource Development
Microbial Ecology of Hydraulic Fracturing: Implications for Sustainable Resource Development Irvine, CA September 13, 2014 Prof. Kelvin B. Gregory Evolution of Oil and Gas Development On the banks of the Youghigheny River, Versailles, PA, 1919. Photo Courtesy: McKeesport Historical Society. Located and Digitized by Prof. Joel Tarr, Carnegie Mellon University. Natural Gas-bearing Shale • Gas trapped in pores or adsorbed to surfaces in low permeability rock. • Commercial production requires engineering permeability • Enabled by: Horizontal Drilling / Hydraulic Fracturing Overview of Horizontal Drilling “Walking Rig” Multiple wells on same pad Horizontal Drilling: Economic and Environmental Advantages • Vertical Wells (pink) many pad sites • Horizontal Well (green) single pad site Centralized Operations, Less Land Disturbance, Lower Construction Costs Hydraulic Fracturing: Water Utilization 1000 m Fracturing Fluid Produced Water returns to contains 4-20 million the surface and is stored liters of water mixed 1500 m prior to treatment and with sand and chemicals reuse, or disposal. that protect well and 2000 m optimize gas production. 2500 m Produced Water Characteristics 1600 250 200 maximum average /d) 1200 3 TDS (mg/L) (m 345,000 106,390 150 Flowrate oil and 800 grease (mg/L) 802 74 TDS 100 Flow rate 400 TOC (mg/L) 1530 160 50 SO4(mg/L) 763 71 TDS Concentration (g/L) Cl (mg/L) 196,000 57,447 0 0 0 50 100 150 Na (mg/L) 117,000 24,123 Time of Flowback (days) Ca (mg/L) 41,000 7,220 Ba (mg/L) 13,800 2,224 Sr (mg/L) 8,460 1,695 Fe total (mg/L) -

Microbial Diversity of Thermophiles with Biomass Deconstruction Potential in a Foliage-Rich Hot Spring
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Universiti Teknologi Malaysia Institutional Repository Received: 9 December 2017 | Revised: 29 January 2018 | Accepted: 12 February 2018 DOI: 10.1002/mbo3.615 ORIGINAL RESEARCH Microbial diversity of thermophiles with biomass deconstruction potential in a foliage- rich hot spring Li Sin Lee1 | Kian Mau Goh2 | Chia Sing Chan2 | Geok Yuan Annie Tan1 | Wai-Fong Yin1 | Chun Shiong Chong2 | Kok-Gan Chan1,3 1ISB (Genetics), Faculty of Science, University of Malaysia, Kuala Abstract Lumpur, Malaysia The ability of thermophilic microorganisms and their enzymes to decompose biomass 2 Faculty of Biosciences and Medical have attracted attention due to their quick reaction time, thermostability, and de- Engineering, Universiti Teknologi Malaysia, Skudai, Johor, Malaysia creased risk of contamination. Exploitation of efficient thermostable glycoside hy- 3Jiangsu University, Zhenjiang, China drolases (GHs) could accelerate the industrialization of biofuels and biochemicals. However, the full spectrum of thermophiles and their enzymes that are important for Correspondence Kok-Gan Chan, International Genome biomass degradation at high temperatures have not yet been thoroughly studied. We Centre, Jiangsu University, Zhenjiang, China. examined a Malaysian Y- shaped Sungai Klah hot spring located within a wooded area. Email: [email protected] The fallen foliage that formed a thick layer of biomass bed under the heated water of Funding information the Y- shaped Sungai Klah hot spring was an ideal environment for the discovery and Postgraduate Research Fund grant, Grant/ Award Number: PG124-2016A; University analysis of microbial biomass decay communities. We sequenced the hypervariable of Malaya, Grant/Award Number: GA001- regions of bacterial and archaeal 16S rRNA genes using total community DNA ex- 2016 and GA002-2016; Universiti Teknologi Malaysia GUP, Grant/Award Number: tracted from the hot spring.