Microbial Metagenomes and Metatranscriptomes During a Coastal Phytoplankton Bloom
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Akashiwo Sanguinea
Ocean ORIGINAL ARTICLE and Coastal http://doi.org/10.1590/2675-2824069.20-004hmdja Research ISSN 2675-2824 Phytoplankton community in a tropical estuarine gradient after an exceptional harmful bloom of Akashiwo sanguinea (Dinophyceae) in the Todos os Santos Bay Helen Michelle de Jesus Affe1,2,* , Lorena Pedreira Conceição3,4 , Diogo Souza Bezerra Rocha5 , Luis Antônio de Oliveira Proença6 , José Marcos de Castro Nunes3,4 1 Universidade do Estado do Rio de Janeiro - Faculdade de Oceanografia (Bloco E - 900, Pavilhão João Lyra Filho, 4º andar, sala 4018, R. São Francisco Xavier, 524 - Maracanã - 20550-000 - Rio de Janeiro - RJ - Brazil) 2 Instituto Nacional de Pesquisas Espaciais/INPE - Rede Clima - Sub-rede Oceanos (Av. dos Astronautas, 1758. Jd. da Granja -12227-010 - São José dos Campos - SP - Brazil) 3 Universidade Estadual de Feira de Santana - Departamento de Ciências Biológicas - Programa de Pós-graduação em Botânica (Av. Transnordestina s/n - Novo Horizonte - 44036-900 - Feira de Santana - BA - Brazil) 4 Universidade Federal da Bahia - Instituto de Biologia - Laboratório de Algas Marinhas (Rua Barão de Jeremoabo, 668 - Campus de Ondina 40170-115 - Salvador - BA - Brazil) 5 Instituto Internacional para Sustentabilidade - (Estr. Dona Castorina, 124 - Jardim Botânico - 22460-320 - Rio de Janeiro - RJ - Brazil) 6 Instituto Federal de Santa Catarina (Av. Ver. Abrahão João Francisco, 3899 - Ressacada, Itajaí - 88307-303 - SC - Brazil) * Corresponding author: [email protected] ABSTRAct The objective of this study was to evaluate variations in the composition and abundance of the phytoplankton community after an exceptional harmful bloom of Akashiwo sanguinea that occurred in Todos os Santos Bay (BTS) in early March, 2007. -

The Closest Unicellular Relatives of Animals
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Current Biology, Vol. 12, 1773–1778, October 15, 2002, 2002 Elsevier Science Ltd. All rights reserved. PII S0960-9822(02)01187-9 The Closest Unicellular Relatives of Animals B.F. Lang,1,2 C. O’Kelly,1,3 T. Nerad,4 M.W. Gray,1,5 Results and Discussion and G. Burger1,2,6 1The Canadian Institute for Advanced Research The evolution of the Metazoa from single-celled protists Program in Evolutionary Biology is an issue that has intrigued biologists for more than a 2 De´ partement de Biochimie century. Early morphological and more recent ultra- Universite´ de Montre´ al structural and molecular studies have converged in sup- Succursale Centre-Ville porting the now widely accepted view that animals are Montre´ al, Que´ bec H3C 3J7 related to Fungi, choanoflagellates, and ichthyosporean Canada protists. However, controversy persists as to the spe- 3 Bigelow Laboratory for Ocean Sciences cific evolutionary relationships among these major P.O. Box 475 groups. This uncertainty is reflected in the plethora of 180 McKown Point Road published molecular phylogenies that propose virtually West Boothbay Harbor, Maine 04575 all of the possible alternative tree topologies involving 4 American Type Culture Collection Choanoflagellata, Fungi, Ichthyosporea, and Metazoa. 10801 University Boulevard For example, a monophyletic MetazoaϩChoanoflagel- Manassas, Virginia 20110 lata group has been suggested on the basis of small 5 Department of Biochemistry subunit (SSU) rDNA sequences [1, 6, 7]. Other studies and Molecular Biology using the same sequences have allied Choanoflagellata Dalhousie University with the Fungi [8], placed Choanoflagellata prior to the Halifax, Nova Scotia B3H 4H7 divergence of animals and Fungi [9], or even placed Canada them prior to the divergence of green algae and land plants [10]. -

The Planktonic Protist Interactome: Where Do We Stand After a Century of Research?
bioRxiv preprint doi: https://doi.org/10.1101/587352; this version posted May 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Bjorbækmo et al., 23.03.2019 – preprint copy - BioRxiv The planktonic protist interactome: where do we stand after a century of research? Marit F. Markussen Bjorbækmo1*, Andreas Evenstad1* and Line Lieblein Røsæg1*, Anders K. Krabberød1**, and Ramiro Logares2,1** 1 University of Oslo, Department of Biosciences, Section for Genetics and Evolutionary Biology (Evogene), Blindernv. 31, N- 0316 Oslo, Norway 2 Institut de Ciències del Mar (CSIC), Passeig Marítim de la Barceloneta, 37-49, ES-08003, Barcelona, Catalonia, Spain * The three authors contributed equally ** Corresponding authors: Ramiro Logares: Institute of Marine Sciences (ICM-CSIC), Passeig Marítim de la Barceloneta 37-49, 08003, Barcelona, Catalonia, Spain. Phone: 34-93-2309500; Fax: 34-93-2309555. [email protected] Anders K. Krabberød: University of Oslo, Department of Biosciences, Section for Genetics and Evolutionary Biology (Evogene), Blindernv. 31, N-0316 Oslo, Norway. Phone +47 22845986, Fax: +47 22854726. [email protected] Abstract Microbial interactions are crucial for Earth ecosystem function, yet our knowledge about them is limited and has so far mainly existed as scattered records. Here, we have surveyed the literature involving planktonic protist interactions and gathered the information in a manually curated Protist Interaction DAtabase (PIDA). In total, we have registered ~2,500 ecological interactions from ~500 publications, spanning the last 150 years. -

Check List 15 (5): 951–963
15 5 ANNOTATED LIST OF SPECIES Check List 15 (5): 951–963 https://doi.org/10.15560/15.5.951 Dinoflagellates in tropical estuarine waters from the Maraú River, Camamu Bay, northeastern Brazil Caio Ceza da Silva Nunes1, Sylvia Maria Moreira Susini-Ribeiro1, 2, Kaoli Pereira Cavalcante3 1 Mestrado em Sistemas Aquáticos Tropicais, Universidade Estadual de Santa Cruz, Rodovia Jorge Amado, km 16, Salobrinho, 45662090 Ilhéus, BA, Brazil. 2 Universidade Estadual de Santa Cruz, Rodovia Jorge Amado, km 16, Salobrinho, 45662090 Ilhéus, BA, Brazil. 3 Universidade Estadual Vale do Acaraú, Avenida da Universidade, 850, Campus da Betânia, Betânia, 62040370, Sobral, CE, Brazil. Corresponding author: Caio Ceza da Silva Nunes, [email protected] Abstract Dinoflagellates display great diversity in tropical regions and play an important role in the complex microbial food webs of marine and brackish environments. The goal of this study is to identify planktonic dinoflagellates and their distribution in the estuary of the Maraú River, Camamu Bay, state of Bahia, in a region with increasing use of shellfish farming. Samples were carried out monthly from August 2006 to July 2007 at four stations along the estuary. Plankton was sampled with a 20 μm mesh net. We identified 20 dinoflagellate species. The greatest species richness was ob- served in the genera Protoperidinium (five spp.), Tripos (four spp.), and Prorocentrum (three spp.). Based on literature, six species were classified as potentially harmful: Akashiwo sanguinea, Dinophysis caudata, Gonyaulax spinifera, Prorocentrum micans, Scrippsiella cf. acuminata, and Tripos furca. Protoperidinium venustum was recorded for the first time in coastal waters of Bahia. Keywords Brackish water, Dinophyta, distribution, potentially harmful species, taxonomy. -

Sex Is a Ubiquitous, Ancient, and Inherent Attribute of Eukaryotic Life
PAPER Sex is a ubiquitous, ancient, and inherent attribute of COLLOQUIUM eukaryotic life Dave Speijera,1, Julius Lukešb,c, and Marek Eliášd,1 aDepartment of Medical Biochemistry, Academic Medical Center, University of Amsterdam, 1105 AZ, Amsterdam, The Netherlands; bInstitute of Parasitology, Biology Centre, Czech Academy of Sciences, and Faculty of Sciences, University of South Bohemia, 370 05 Ceské Budejovice, Czech Republic; cCanadian Institute for Advanced Research, Toronto, ON, Canada M5G 1Z8; and dDepartment of Biology and Ecology, University of Ostrava, 710 00 Ostrava, Czech Republic Edited by John C. Avise, University of California, Irvine, CA, and approved April 8, 2015 (received for review February 14, 2015) Sexual reproduction and clonality in eukaryotes are mostly Sex in Eukaryotic Microorganisms: More Voyeurs Needed seen as exclusive, the latter being rather exceptional. This view Whereas absence of sex is considered as something scandalous for might be biased by focusing almost exclusively on metazoans. a zoologist, scientists studying protists, which represent the ma- We analyze and discuss reproduction in the context of extant jority of extant eukaryotic diversity (2), are much more ready to eukaryotic diversity, paying special attention to protists. We accept that a particular eukaryotic group has not shown any evi- present results of phylogenetically extended searches for ho- dence of sexual processes. Although sex is very well documented mologs of two proteins functioning in cell and nuclear fusion, in many protist groups, and members of some taxa, such as ciliates respectively (HAP2 and GEX1), providing indirect evidence for (Alveolata), diatoms (Stramenopiles), or green algae (Chlor- these processes in several eukaryotic lineages where sex has oplastida), even serve as models to study various aspects of sex- – not been observed yet. -

University of Oklahoma
UNIVERSITY OF OKLAHOMA GRADUATE COLLEGE MACRONUTRIENTS SHAPE MICROBIAL COMMUNITIES, GENE EXPRESSION AND PROTEIN EVOLUTION A DISSERTATION SUBMITTED TO THE GRADUATE FACULTY in partial fulfillment of the requirements for the Degree of DOCTOR OF PHILOSOPHY By JOSHUA THOMAS COOPER Norman, Oklahoma 2017 MACRONUTRIENTS SHAPE MICROBIAL COMMUNITIES, GENE EXPRESSION AND PROTEIN EVOLUTION A DISSERTATION APPROVED FOR THE DEPARTMENT OF MICROBIOLOGY AND PLANT BIOLOGY BY ______________________________ Dr. Boris Wawrik, Chair ______________________________ Dr. J. Phil Gibson ______________________________ Dr. Anne K. Dunn ______________________________ Dr. John Paul Masly ______________________________ Dr. K. David Hambright ii © Copyright by JOSHUA THOMAS COOPER 2017 All Rights Reserved. iii Acknowledgments I would like to thank my two advisors Dr. Boris Wawrik and Dr. J. Phil Gibson for helping me become a better scientist and better educator. I would also like to thank my committee members Dr. Anne K. Dunn, Dr. K. David Hambright, and Dr. J.P. Masly for providing valuable inputs that lead me to carefully consider my research questions. I would also like to thank Dr. J.P. Masly for the opportunity to coauthor a book chapter on the speciation of diatoms. It is still such a privilege that you believed in me and my crazy diatom ideas to form a concise chapter in addition to learn your style of writing has been a benefit to my professional development. I’m also thankful for my first undergraduate research mentor, Dr. Miriam Steinitz-Kannan, now retired from Northern Kentucky University, who was the first to show the amazing wonders of pond scum. Who knew that studying diatoms and algae as an undergraduate would lead me all the way to a Ph.D. -

Protocols for Monitoring Harmful Algal Blooms for Sustainable Aquaculture and Coastal Fisheries in Chile (Supplement Data)
Protocols for monitoring Harmful Algal Blooms for sustainable aquaculture and coastal fisheries in Chile (Supplement data) Provided by Kyoko Yarimizu, et al. Table S1. Phytoplankton Naming Dictionary: This dictionary was constructed from the species observed in Chilean coast water in the past combined with the IOC list. Each name was verified with the list provided by IFOP and online dictionaries, AlgaeBase (https://www.algaebase.org/) and WoRMS (http://www.marinespecies.org/). The list is subjected to be updated. Phylum Class Order Family Genus Species Ochrophyta Bacillariophyceae Achnanthales Achnanthaceae Achnanthes Achnanthes longipes Bacillariophyta Coscinodiscophyceae Coscinodiscales Heliopeltaceae Actinoptychus Actinoptychus spp. Dinoflagellata Dinophyceae Gymnodiniales Gymnodiniaceae Akashiwo Akashiwo sanguinea Dinoflagellata Dinophyceae Gymnodiniales Gymnodiniaceae Amphidinium Amphidinium spp. Ochrophyta Bacillariophyceae Naviculales Amphipleuraceae Amphiprora Amphiprora spp. Bacillariophyta Bacillariophyceae Thalassiophysales Catenulaceae Amphora Amphora spp. Cyanobacteria Cyanophyceae Nostocales Aphanizomenonaceae Anabaenopsis Anabaenopsis milleri Cyanobacteria Cyanophyceae Oscillatoriales Coleofasciculaceae Anagnostidinema Anagnostidinema amphibium Anagnostidinema Cyanobacteria Cyanophyceae Oscillatoriales Coleofasciculaceae Anagnostidinema lemmermannii Cyanobacteria Cyanophyceae Oscillatoriales Microcoleaceae Annamia Annamia toxica Cyanobacteria Cyanophyceae Nostocales Aphanizomenonaceae Aphanizomenon Aphanizomenon flos-aquae -

Biology and Systematics of Heterokont and Haptophyte Algae1
American Journal of Botany 91(10): 1508±1522. 2004. BIOLOGY AND SYSTEMATICS OF HETEROKONT AND HAPTOPHYTE ALGAE1 ROBERT A. ANDERSEN Bigelow Laboratory for Ocean Sciences, P.O. Box 475, West Boothbay Harbor, Maine 04575 USA In this paper, I review what is currently known of phylogenetic relationships of heterokont and haptophyte algae. Heterokont algae are a monophyletic group that is classi®ed into 17 classes and represents a diverse group of marine, freshwater, and terrestrial algae. Classes are distinguished by morphology, chloroplast pigments, ultrastructural features, and gene sequence data. Electron microscopy and molecular biology have contributed signi®cantly to our understanding of their evolutionary relationships, but even today class relationships are poorly understood. Haptophyte algae are a second monophyletic group that consists of two classes of predominately marine phytoplankton. The closest relatives of the haptophytes are currently unknown, but recent evidence indicates they may be part of a large assemblage (chromalveolates) that includes heterokont algae and other stramenopiles, alveolates, and cryptophytes. Heter- okont and haptophyte algae are important primary producers in aquatic habitats, and they are probably the primary carbon source for petroleum products (crude oil, natural gas). Key words: chromalveolate; chromist; chromophyte; ¯agella; phylogeny; stramenopile; tree of life. Heterokont algae are a monophyletic group that includes all (Phaeophyceae) by Linnaeus (1753), and shortly thereafter, photosynthetic organisms with tripartite tubular hairs on the microscopic chrysophytes (currently 5 Oikomonas, Anthophy- mature ¯agellum (discussed later; also see Wetherbee et al., sa) were described by MuÈller (1773, 1786). The history of 1988, for de®nitions of mature and immature ¯agella), as well heterokont algae was recently discussed in detail (Andersen, as some nonphotosynthetic relatives and some that have sec- 2004), and four distinct periods were identi®ed. -

Multigene Eukaryote Phylogeny Reveals the Likely Protozoan Ancestors of Opis- Thokonts (Animals, Fungi, Choanozoans) and Amoebozoa
Accepted Manuscript Multigene eukaryote phylogeny reveals the likely protozoan ancestors of opis- thokonts (animals, fungi, choanozoans) and Amoebozoa Thomas Cavalier-Smith, Ema E. Chao, Elizabeth A. Snell, Cédric Berney, Anna Maria Fiore-Donno, Rhodri Lewis PII: S1055-7903(14)00279-6 DOI: http://dx.doi.org/10.1016/j.ympev.2014.08.012 Reference: YMPEV 4996 To appear in: Molecular Phylogenetics and Evolution Received Date: 24 January 2014 Revised Date: 2 August 2014 Accepted Date: 11 August 2014 Please cite this article as: Cavalier-Smith, T., Chao, E.E., Snell, E.A., Berney, C., Fiore-Donno, A.M., Lewis, R., Multigene eukaryote phylogeny reveals the likely protozoan ancestors of opisthokonts (animals, fungi, choanozoans) and Amoebozoa, Molecular Phylogenetics and Evolution (2014), doi: http://dx.doi.org/10.1016/ j.ympev.2014.08.012 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. 1 1 Multigene eukaryote phylogeny reveals the likely protozoan ancestors of opisthokonts 2 (animals, fungi, choanozoans) and Amoebozoa 3 4 Thomas Cavalier-Smith1, Ema E. Chao1, Elizabeth A. Snell1, Cédric Berney1,2, Anna Maria 5 Fiore-Donno1,3, and Rhodri Lewis1 6 7 1Department of Zoology, University of Oxford, South Parks Road, Oxford OX1 3PS, UK. -
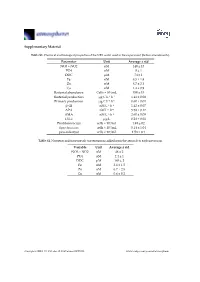
Supplementary Material Parameter Unit Average ± Std NO3 + NO2 Nm
Supplementary Material Table S1. Chemical and biological properties of the NRS water used in the experiment (before amendments). Parameter Unit Average ± std NO3 + NO2 nM 140 ± 13 PO4 nM 8 ± 1 DOC μM 74 ± 1 Fe nM 8.5 ± 1.8 Zn nM 8.7 ± 2.1 Cu nM 1.4 ± 0.9 Bacterial abundance Cells × 104/mL 350 ± 15 Bacterial production μg C L−1 h−1 1.41 ± 0.08 Primary production μg C L−1 h−1 0.60 ± 0.01 β-Gl nM L−1 h−1 1.42 ± 0.07 APA nM L−1 h−1 5.58 ± 0.17 AMA nM L−1·h−1 2.60 ± 0.09 Chl-a μg/L 0.28 ± 0.01 Prochlorococcus cells × 104/mL 1.49 ± 02 Synechococcus cells × 104/mL 5.14 ± 1.04 pico-eukaryot cells × 103/mL 1.58 × 0.1 Table S2. Nutrients and trace metals concentrations added from the aerosols to each mesocosm. Variable Unit Average ± std NO3 + NO2 nM 48 ± 2 PO4 nM 2.4 ± 1 DOC μM 165 ± 2 Fe nM 2.6 ± 1.5 Zn nM 6.7 ± 2.5 Cu nM 0.6 ± 0.2 Atmosphere 2019, 10, 358; doi:10.3390/atmos10070358 www.mdpi.com/journal/atmosphere Atmosphere 2019, 10, 358 2 of 6 Table S3. ANOVA test results between control, ‘UV-treated’ and ‘live-dust’ treatments at 20 h or 44 h, with significantly different values shown in bold. ANOVA df Sum Sq Mean Sq F Value p-value Chl-a 20 H 2, 6 0.03, 0.02 0.02, 0 4.52 0.0634 44 H 2, 6 0.02, 0 0.01, 0 23.13 0.002 Synechococcus Abundance 20 H 2, 7 8.23 × 107, 4.11 × 107 4.11 × 107, 4.51 × 107 0.91 0.4509 44 H 2, 7 5.31 × 108, 6.97 × 107 2.65 × 108, 1.16 × 107 22.84 0.0016 Prochlorococcus Abundance 20 H 2, 8 4.22 × 107, 2.11 × 107 2.11 × 107, 2.71 × 106 7.77 0.0216 44 H 2, 8 9.02 × 107, 1.47 × 107 4.51 × 107, 2.45 × 106 18.38 0.0028 Pico-eukaryote -

Heterotrophic ¯ Agellates (Protista) from Marine Sediments of Botany Bay, Australia
Journal of Natural History, 2000, 34, 483± 562 Heterotrophic ¯ agellates (Protista) from marine sediments of Botany Bay, Australia WON JE LEE and DAVID J. PATTERSON School of Biological Sciences, University of Sydney, NSW 2006, Australia; e-mail: [email protected] (Accepted 19 April 1999) Heterotrophic¯ agellates (protozoa) occurring in the marine sediments at Botany Bay, Australia are reported. Among the 87 species from 43 genera encountered in this survey are 13 new taxa: Cercomonas granulatus n. sp., Clautriavia cavus n. sp., Heteronema larseni n. sp., Notosolenus adamas n. sp., Notosolenus brothernis n. sp., Notosolenus hemicircularis n. sp., Notosolenus lashue n. sp., Notosolenus pyriforme n. sp., Petalomonas intortus n. sp., Petalomonas iugosus n. sp., Petalomonas labrum n. sp., Petalomonas planus n. sp. and Petalomonas virgatus n. sp.; and seven new combinations, Carpediemonas bialata n. comb., Dinema platysomum n. comb., Petalomonas calycimonoides nom. nov., Petalomonas chris- teni nom. nov., Petalomonas physaloides n. comb., Petalomonas quinquecarinata n. comb. and Petalomonas spinifera n. comb. Most ¯ agellates described here appear to be cosmopolitan.We are unable to assess if the new species are endemic because of the lack of intensive studies elsewhere. Keywords: Biogeography, endemic biota, heterotrophic ¯ agellates, taxonomy, protists. Introduction Marine heterotrophic protists are predators on bacteria and small phytoplankton, are prey for larger zooplankton, and facilitate remineralization and recycling of elements essential for phytoplankton and microbial growth (Azam et al., 1983; Porter et al., 1985; Sherr and Sherr, 1988; JuÈ rgens and GuÈ de, 1990; Kirchman, 1994; Pace and Vaque , 1994). Consequently, the role of heterotrophic protists in planktonic microbial food webs of marine environments has received increasing attention. -

Predatory Flagellates – the New Recently Discovered Deep Branches of the Eukaryotic Tree and Their Evolutionary and Ecological Significance
Protistology 14 (1), 15–22 (2020) Protistology Predatory flagellates – the new recently discovered deep branches of the eukaryotic tree and their evolutionary and ecological significance Denis V. Tikhonenkov Papanin Institute for Biology of Inland Waters, Russian Academy of Sciences, Borok, 152742, Russia | Submitted March 20, 2020 | Accepted April 6, 2020 | Summary Predatory protists are poorly studied, although they are often representing important deep-branching evolutionary lineages and new eukaryotic supergroups. This short review/opinion paper is inspired by the recent discoveries of various predatory flagellates, which form sister groups of the giant eukaryotic clusters on phylogenetic trees, and illustrate an ancestral state of one or another supergroup of eukaryotes. Here we discuss their evolutionary and ecological relevance and show that the study of such protists may be essential in addressing previously puzzling evolutionary problems, such as the origin of multicellular animals, the plastid spread trajectory, origins of photosynthesis and parasitism, evolution of mitochondrial genomes. Key words: evolution of eukaryotes, heterotrophic flagellates, mitochondrial genome, origin of animals, photosynthesis, predatory protists, tree of life Predatory flagellates and diversity of eu- of the hidden diversity of protists (Moon-van der karyotes Staay et al., 2000; López-García et al., 2001; Edg- comb et al., 2002; Massana et al., 2004; Richards The well-studied multicellular animals, plants and Bass, 2005; Tarbe et al., 2011; de Vargas et al., and fungi immediately come to mind when we hear 2015). In particular, several prevailing and very abun- the term “eukaryotes”. However, these groups of dant ribogroups such as MALV, MAST, MAOP, organisms represent a minority in the real diversity MAFO (marine alveolates, stramenopiles, opistho- of evolutionary lineages of eukaryotes.