Derived Neural Progenitor Cells Reveal Early Neurodevelopmental Phenotype for Koolen-De Vries Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DNA Insertion Mutations Can Be Predicted by a Periodic Probability
Research article DNA insertion mutations can be predicted by a periodic probability function Tatsuaki Tsuruyama1 1Department of Pathology, Graduate School of Medicine, Kyoto University, Yoshida Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan Running title: Probability prediction of mutation Correspondence to: Tatsuaki Tsuruyama Kyoto University, Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan Tel.: +81-75-751-3488; Fax: +81-75-761-9591 E-mail: [email protected] 1 Abstract It is generally difficult to predict the positions of mutations in genomic DNA at the nucleotide level. Retroviral DNA insertion is one mode of mutation, resulting in host infections that are difficult to treat. This mutation process involves the integration of retroviral DNA into the host-infected cellular genomic DNA following the interaction between host DNA and a pre-integration complex consisting of retroviral DNA and integrase. Here, we report that retroviral insertion sites around a hotspot within the Zfp521 and N-myc genes can be predicted by a periodic function that is deduced using the diffraction lattice model. In conclusion, the mutagenesis process is described by a biophysical model for DNA–DNA interactions. Keywords: Insertion, mutagenesis, palindromic sequence, retroviral DNA 2 Text Introduction Extensive research has examined retroviral insertions to further our understanding of DNA mutations. Retrovirus-related diseases, including leukemia/lymphoma and AIDS, develop after retroviral genome insertion into the genomic DNA of the infected host cell. Retroviral DNA insertion is one of the modes of insertional mutation. After reverse-transcription of the retroviral genomic RNA into DNA, the retroviral DNA forms a pre-insertion complex (PIC) with the integrase enzyme, which catalyzes the insertion reaction. -

DNA Sequence Insertion and Evolutionary Variation in Gene Regulation (Mobile Elements/Long Terminal Repeats/Alu Sequences/Factor-Binding Sites) Roy J
Proc. Natl. Acad. Sci. USA Vol. 93, pp. 9374-9377, September 1996 Colloquium Paper This paper was presented at a colloquium entitled "Biology of Developmental Transcription Control, " organized by Eric H. Davidson, Roy J. Britten, and Gary Felsenfeld, held October 26-28, 1995, at the National Academy of Sciences in Irvine, CA. DNA sequence insertion and evolutionary variation in gene regulation (mobile elements/long terminal repeats/Alu sequences/factor-binding sites) RoY J. BRITrEN Division of Biology, California Institute of Technology, 101 Dahlia Avenue, Corona del Mar, CA 92625 ABSTRACT Current evidence on the long-term evolution- 3 and 4). Sequence change, obscuring the original structure, ary effect of insertion of sequence elements into gene regions has occurred in the long history, and the underlying rate of is reviewed, restricted to cases where a sequence derived from base substitution that is responsible is known (5). a past insertion participates in the regulation of expression of The requirements for a convincing example are: (i) that a useful gene. Ten such examples in eukaryotes demonstrate there be a trace of a known class of elements present in gene that segments of repetitive DNA or mobile elements have been region; (ii) that there is evidence that it has been there long inserted in the past in gene regions, have been preserved, enough to not just be a transient mutation; (iii) that some sometimes modified by selection, and now affect control of sequence residue of the mobile element or repeat participates transcription ofthe adjacent gene. Included are only examples in regulation of expression of the gene; (iv) that the gene have in which transcription control was modified by the insert. -

Genetic Analysis of the Transition from Wild to Domesticated Cotton (G
bioRxiv preprint doi: https://doi.org/10.1101/616763; this version posted April 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genetic analysis of the transition from wild to domesticated cotton (G. hirsutum) Corrinne E. Grover1, Mi-Jeong Yoo1,a, Michael A. Gore2, David B. Harker3,b, Robert L. Byers3, Alexander E. Lipka4, Guanjing Hu1, Daojun Yuan1, Justin L. Conover1, Joshua A. Udall3,c, Andrew H. Paterson5, and Jonathan F. Wendel1 1Department of Ecology, Evolution and Organismal Biology, Iowa State University, Ames, IA 50011 2Plant Breeding and Genetics Section, School of Integrative Plant Science, Cornell University, Ithaca, NY 14853 3Plant and Wildlife Science Department, Brigham Young University, Provo, UT 84602 4Department of Crop Sciences, University of Illinois, Urbana, IL 61801 5Plant Genome Mapping Laboratory, University of Georgia, Athens, GA 30602 acurrent address: Department of Biology, University of Florida, Gainesville, FL 32611 bcurrent address: Department of Dermatology, The University of Texas Southwestern Medical Center, Dallas, TX 75390 ccurrent address: Southern Plains Agricultural Research Center, USDA-ARS, College Station, TX, 77845- 4988 Corresponding Author: Jonathan F. Wendel ([email protected]) Data Availability: All data are available via figshare (https://figshare.com/projects/Genetic_analysis_of_the_transition_from_wild_to_domesticated_cotton_G _hirsutum_QTL/62693) and GitHub (https://github.com/Wendellab/QTL_TxMx). Keywords: QTL, domestication, Gossypium hirsutum, cotton Summary: An F2 population between truly wild and domesticated cotton was used to identify QTL associated with selection under domestication. -

Insertion Element (IS1 Insertion Sequence/Chloramphenicol Resistance Transposon Tn9/Integrative Recombination) L
Proc. Nati. Acad. Sci. USA Vol. 75, No. 3, pp. 1490-1494, March 1978 Genetics Chromosomal integration of phage X by means of a DNA insertion element (IS1 insertion sequence/chloramphenicol resistance transposon Tn9/integrative recombination) L. A. MACHATTIE AND J. A. SHAPIRO Department of Microbiology, University of Chicago, Chicago, Illinois 60637 Communicated by Albert Dorfman, January 10, 1978 ABSTRACT Phage Xcamll2, which contains the chlor- carries a deletion of the gal-attB-bio region of the Escherichia amphenicol resistance transposon Tn9 and has a deletion of attP coil chromosome. MGBO is a gal+bio+ transductant of and the int gene, will lysogenize Escherichia coli K-12. Pro- phage integration occurs at different chromosomal sites, in- MADO. MS6 is a galE indicator for detection of XgalE + T- cluding lacYand maiB, but not at attB. All Xcamll2 prophages transducing particles and S1653 is a gal deletion strain for de- are excised from the chromosome after induction but with tection of Xgal + particles (3). Strain 200PS is a thi lacY strain various efficiencies for different locations. Heteroduplex from the Pasteur collection. Strain QL carries a complete lac analysis of XplacZ transducing phages isolated from a lacY:: deletion, strain X9003 carries the nonpolar M15 lacZ deletion, Xcamll2 prophage reveals an insertion sequence 1 (IS1) element and either will serve as indicator for XplacZ phage in a blue at theloint of viral and chromosomal DNA. Two lines of evi- dencekdicate that Xcamll2 encodes an excision activity that plaque assay (8). recognizes the ISI element: (i) prophage derepression increases Media. Our basic minimal medium and complete TYE the frequency of excision from IacYto yield lac+ revertants, and medium have been described (9). -

Gene Therapy Glossary of Terms
GENE THERAPY GLOSSARY OF TERMS A • Phase 3: A phase of research to describe clinical trials • Allele: one of two or more alternative forms of a gene that that gather more information about a drug’s safety and arise by mutation and are found at the same place on a effectiveness by studying different populations and chromosome. different dosages and by using the drug in combination • Adeno-Associated Virus: A single stranded DNA virus that has with other drugs. These studies typically involve more not been found to cause disease in humans. This type of virus participants.7 is the most frequently used in gene therapy.1 • Phase 4: A phase of research to describe clinical trials • Adenovirus: A member of a family of viruses that can cause occurring after FDA has approved a drug for marketing. infections in the respiratory tract, eye, and gastrointestinal They include post market requirement and commitment tract. studies that are required of or agreed to by the study • Adeno-Associated Virus Vector: Adeno viruses used as sponsor. These trials gather additional information about a vehicles for genes, whose core genetic material has been drug’s safety, efficacy, or optimal use.8 removed and replaced by the FVIII- or FIX-gene • Codon: a sequence of three nucleotides in DNA or RNA • Amino Acids: building block of a protein that gives instructions to add a specific amino acid to an • Antibody: a protein produced by immune cells called B-cells elongating protein in response to a foreign molecule; acts by binding to the • CRISPR: a family of DNA sequences that can be cleaved by molecule and often making it inactive or targeting it for specific enzymes, and therefore serve as a guide to cut out destruction and insert genes. -

Genetic Testing: Background and Policy Issues
Genetic Testing: Background and Policy Issues Amanda K. Sarata Specialist in Health Policy March 2, 2015 Congressional Research Service 7-5700 www.crs.gov RL33832 Genetic Testing: Background and Policy Issues Summary Congress has considered, at various points in time, numerous pieces of legislation that relate to genetic and genomic technology and testing. These include bills addressing genetic discrimination in health insurance and employment; precision medicine; the patenting of genetic material; and the oversight of clinical laboratory tests (in vitro diagnostics), including genetic tests. The focus on these issues signals the growing importance of public policy issues surrounding the clinical and public health implications of new genetic technology. As genetic technologies proliferate and are increasingly used to guide clinical treatment, these public policy issues are likely to continue to garner attention. Understanding the basic scientific concepts underlying genetics and genetic testing may help facilitate the development of more effective public policy in this area. Humans have 23 pairs of chromosomes in the nucleus of most cells in their bodies. Chromosomes are composed of deoxyribonucleic acid (DNA) and protein. DNA is composed of complex chemical substances called bases. Proteins are fundamental components of all living cells, and include enzymes, structural elements, and hormones. A gene is the section of DNA that contains the sequence which corresponds to a specific protein. Though most of the genome is similar between individuals, there can be significant variation in physical appearance or function between individuals due to variations in DNA sequence that may manifest as changes in the protein, which affect the protein’s function. -

The Origin, Evolution, and Functional Impact of Short Insertion–Deletion Variants Identified in 179 Human Genomes
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Research The origin, evolution, and functional impact of short insertion–deletion variants identified in 179 human genomes Stephen B. Montgomery,1,2,3,14,16 David L. Goode,3,14,15 Erika Kvikstad,4,13,14 Cornelis A. Albers,5,6 Zhengdong D. Zhang,7 Xinmeng Jasmine Mu,8 Guruprasad Ananda,9 Bryan Howie,10 Konrad J. Karczewski,3 Kevin S. Smith,2 Vanessa Anaya,2 Rhea Richardson,2 Joe Davis,3 The 1000 Genomes Pilot Project Consortium, Daniel G. MacArthur,5,11 Arend Sidow,2,3 Laurent Duret,4 Mark Gerstein,8 Kateryna D. Makova,9 Jonathan Marchini,12 Gil McVean,12,13 and Gerton Lunter13,16 1–13[Author affiliations appear at the end of the paper.] Short insertions and deletions (indels) are the second most abundant form of human genetic variation, but our un- derstanding of their origins and functional effects lags behind that of other types of variants. Using population-scale sequencing, we have identified a high-quality set of 1.6 million indels from 179 individuals representing three diverse human populations. We show that rates of indel mutagenesis are highly heterogeneous, with 43%–48% of indels occurring in 4.03% of the genome, whereas in the remaining 96% their prevalence is 16 times lower than SNPs. Polymerase slippage can explain upwards of three-fourths of all indels, with the remainder being mostly simple de- letions in complex sequence. However, insertions do occur and are significantly associated with pseudo-palindromic sequence features compatible with the fork stalling and template switching (FoSTeS) mechanism more commonly as- sociated with large structural variations. -

Characterization of Point Mutations in the Same Arginine Codon in Three Unrelated Patients with Ornithine Transcarbamylase Deficiency
Characterization of point mutations in the same arginine codon in three unrelated patients with ornithine transcarbamylase deficiency. A Maddalena, … , W E O'Brien, R L Nussbaum J Clin Invest. 1988;82(4):1353-1358. https://doi.org/10.1172/JCI113738. Research Article Point mutations in the X-linked ornithine transcarbamylase (OTC) gene have been detected at the same Taq I restriction site in 3 of 24 unrelated probands with OTC deficiency. A de novo mutation could be traced in all three families to an individual in a prior generation, confirming independent recurrence. The DNA sequence in the region of the altered Taq I site was determined in the three probands. In two unrelated male probands with neonatal onset of severe OTC deficiency, a guanine (G) to adenine (A) mutation on the sense strand (antisense cytosine [C] to thymine [T]) was found, resulting in glutamine for arginine at amino acid 109 of the mature polypeptide. In the third case, where the proband was a symptomatic female, C to T (sense strand) transition converted residue 109 to a premature stop. These results support the observation that Taq I restriction sites, which contain an internal CG, are particularly susceptible to C to T transition mutation due to deamination of a methylated C in either the sense or antisense strand. The OTC gene seems especially sensitive to C to T transition mutation at arginine codon 109 because either a nonsense mutation or an extremely deleterious missense mutation will result. Find the latest version: https://jci.me/113738/pdf Characterization of Point Mutations in the Same Arginine Codon in Three Unrelated Patients with Ornithine Transcarbamylase Deficiency Anne Maddalena,*" J. -

Glossary of Common Terms in Genetics
Glossary of Common Terms in Genetics Acquired mutations Gene changes genetic information. DNA is held Multiplexing A sequencing approach that that arise within individual cells and together by weak bonds between base uses several pooled samples simultaneous accumulate throughout a person's life pairs of nucleotides: adenine, guanine, ly, greatly increasing sequencing speed. span. cytosine, and thymine. Mutation Any heritable change in DNA Alleles One of a group of genes that Gene The fundamental unit of heredi sequence. occur alternatively at a given locus. A ty. A gene is an ordered sequence of single allele is inherited separately from nucleotides located in a particular posi Nucleotide A subunit of DNA or RNA each parent (e.g., at a locus for eye tion on a particular chromosome that consisting of a nitrogenous base, a phos color, the allele might result in blue or encodes a specific functional product phate molecule, and a sugar molecule. brown eyes). (i.e., a protein or RNA molecule i. Thousands of nucleotides are linked to form a DNA or RNA molecule. Base pair Two nitrogenous bases (ade Gene expression The process by which nine and thymine or guanine and cyto- a gene's coded information is converted Oncogene One or more forms of a sine) held together by weak bonds. Two into the structures present and operat gene associated with cancer. strands of DNA are held together in the ing in the cell. shape of a double helix by the bonds Polygenic disorders Genetic disorders between base pairs. Gene mapping Determination of the resulting from the combined action of relative positions of genes on a DNA alleles of more than one gene (e.g., Carrier A person who has a recessive molecule and the distance between heart disease, diabetes, and some can mutated gene along with its normal them. -

Indel Information Eliminates Trivial Sequence Alignment in Maximum Likelihood Phylogenetic Analysis
Cladistics Cladistics 28 (2012) 514–528 10.1111/j.1096-0031.2012.00402.x Indel information eliminates trivial sequence alignment in maximum likelihood phylogenetic analysis John S.S. Dentona,b,* and Ward C. Wheelerb,c aDivision of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA; bRichard Gilder Graduate School, American Museum of Natural History, New York, NY 10024, USA; cDivision of Invertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA Accepted 12 March 2012 Abstract Although there has been a recent proliferation in maximum-likelihood (ML)-based tree estimation methods based on a fixed sequence alignment (MSA), little research has been done on incorporating indel information in this traditional framework. We show, using a simple model on a single character example, that a trivial alignment of a different form than that previously identified for parsimony is optimal in ML under standard assumptions treating indels as ‘‘missing’’ data, but that it is not optimal when indels are incorporated into the character alphabet. We show that the optimality of the trivial alignment is not an artefact of simplified theory assumptions by demonstrating that trivial alignment likelihoods of five different multiple sequence alignment datasets exhibit this phenomenon. These results demonstrate the need for use of indel information in likelihood analysis on fixed MSAs, and suggest that caution must be exercised when drawing conclusions from software implementations claiming improvements in likelihood scores under an indels-as-missing assumption. Ó The Willi Hennig Society 2012. Maximum likelihood (ML) has become a popular the increasing sophistication of search heuristics in ML optimality criterion for inferring evolutionary trees software (e.g. -
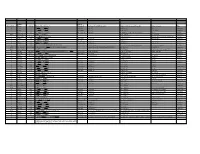
Systematic (Genomic Based)
Trivial Codon Exon/Intron Name Codon Nucleotide Change* Changed Systematic(cDNA based) Systematic (genomic based) Other names Type 2 M-1T -1 CCATGGC>CCACGGC Met>Thr c.2T>C g.4895T>C M1T transition 2 R3Op 3 CACCGAT>CACTGAT Arg>Opal c.10C>T g.4903C>T R3X, R3ter transition 2 ∆A20 20 CCC[A]GAG>CCCGAG Gln>Arg (fs) c.62delA g.4955delA (first base of deletion) ∆A20 deletion IVS2 IVS2-1G>A atagGTA>ataaGTA g.5814G>A c.113-1G>A transition 3 IVS2-1∆4 37-38 ata[gGTA]CCA>ataCCA g.5813delGGTA G12/∆3;IVS2-delGGTA;∆4IVS2-E3 deletion 3 R59op 59 TTCCGAG>TTCTGAG Arg>Opal c.178C>T g.5880C>T R59X;R59ter transition 3 I73T 73 GCATCGG>GCACCGG Ile>Thr c.221T>C g.5923T>C transition 3 L83∆C 83 CCT[C]TAC>CCTTAC Leu>Leu(fs) c.252delC g.5954delC c.250 delC deletion 3 1123ins12 104 GGT[]GGG>GGT[GGGGATCGTGGT]GGG c.314^315ins12 g.6016^6017insGGGGATCGTGGT —12E3;V104+GIVV insertion IVS3 K107K∆12 104-107del GTG[GTGGGAATCAAG]gtt>GTGgtgggaatcaaagtt c.324G>A;delGTGGGAATCAAG(312-324) g.6026G>A g.1133G>A transition IVS3 IVS3-1G>A acagTTA>acaaTTA g.7257G>A c.325-1G>C, g.8180G>C,IVS3sas transversion 4 ∆28E4 114-151 TCC[TCTTGCAGGAACAAACAAAGAAACCACC]ATT>TCCATT Pro>Pro(fs) c.345-372del28 g.7277^7305del c.345_72del28 deletion 4 Q110Oc 110 GACCAAG>GACTAAG Gln>Ochre c.331C>T g.7264C>T transition 4 ∆4E4 118-120 GAAC[AAAC]AAAG>GAACAAAG c.357delAAAC g.7289delAAAC ∆4, MD∆4 deletion 4-5 ∆E4-E5 ccc[tgt…TCC]AGC>cccAGC g.6594^8239del F13/∆4,5 deletion 5 C134R 134 CGCTGTG>CGCCGTG Cys>Arg c.403T>C g.8162T>C C135R transition 5 W147R 147 AAGTGGC>AAGCGGC Trp>Arg c.442T>C g.8201T>C W148R transition -

G14TBS Part II: Population Genetics
G14TBS Part II: Population Genetics Dr Richard Wilkinson Room C26, Mathematical Sciences Building Please email corrections and suggestions to [email protected] Spring 2015 2 Preliminaries These notes form part of the lecture notes for the mod- ule G14TBS. This section is on Population Genetics, and contains 14 lectures worth of material. During this section will be doing some problem-based learning, where the em- phasis is on you to work with your classmates to generate your own notes. I will be on hand at all times to answer questions and guide the discussions. Reading List: There are several good introductory books on population genetics, although I found their level of math- ematical sophistication either too low or too high for the purposes of this module. The following are the sources I used to put together these lecture notes: • Gillespie, J. H., Population genetics: a concise guide. John Hopkins University Press, Baltimore and Lon- don, 2004. • Hartl, D. L., A primer of population genetics, 2nd edition. Sinauer Associates, Inc., Publishers, 1988. • Ewens, W. J., Mathematical population genetics: (I) Theoretical Introduction. Springer, 2000. • Holsinger, K. E., Lecture notes in population genet- ics. University of Connecticut, 2001-2010. Available online at http://darwin.eeb.uconn.edu/eeb348/lecturenotes/notes.html. • Tavar´eS., Ancestral inference in population genetics. In: Lectures on Probability Theory and Statistics. Ecole d'Et´esde Probabilit´ede Saint-Flour XXXI { 2001. (Ed. Picard J.) Lecture Notes in Mathematics, Springer Verlag, 1837, 1-188, 2004. Available online at http://www.cmb.usc.edu/people/stavare/STpapers-pdf/T04.pdf Gillespie, Hartl and Holsinger are written for non-mathematicians and are easiest to follow.