Severe Childhood Speech Disorder Gene Discovery Highlights Transcriptional Dysregulation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of the Transition from Wild to Domesticated Cotton (G
bioRxiv preprint doi: https://doi.org/10.1101/616763; this version posted April 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genetic analysis of the transition from wild to domesticated cotton (G. hirsutum) Corrinne E. Grover1, Mi-Jeong Yoo1,a, Michael A. Gore2, David B. Harker3,b, Robert L. Byers3, Alexander E. Lipka4, Guanjing Hu1, Daojun Yuan1, Justin L. Conover1, Joshua A. Udall3,c, Andrew H. Paterson5, and Jonathan F. Wendel1 1Department of Ecology, Evolution and Organismal Biology, Iowa State University, Ames, IA 50011 2Plant Breeding and Genetics Section, School of Integrative Plant Science, Cornell University, Ithaca, NY 14853 3Plant and Wildlife Science Department, Brigham Young University, Provo, UT 84602 4Department of Crop Sciences, University of Illinois, Urbana, IL 61801 5Plant Genome Mapping Laboratory, University of Georgia, Athens, GA 30602 acurrent address: Department of Biology, University of Florida, Gainesville, FL 32611 bcurrent address: Department of Dermatology, The University of Texas Southwestern Medical Center, Dallas, TX 75390 ccurrent address: Southern Plains Agricultural Research Center, USDA-ARS, College Station, TX, 77845- 4988 Corresponding Author: Jonathan F. Wendel ([email protected]) Data Availability: All data are available via figshare (https://figshare.com/projects/Genetic_analysis_of_the_transition_from_wild_to_domesticated_cotton_G _hirsutum_QTL/62693) and GitHub (https://github.com/Wendellab/QTL_TxMx). Keywords: QTL, domestication, Gossypium hirsutum, cotton Summary: An F2 population between truly wild and domesticated cotton was used to identify QTL associated with selection under domestication. -

8. Eligibility for Special Education Services A
8. Eligibility for Special Education Services a. Fact Sheets on i. ADHD Fact Sheet on Disabilities from NICHCY (http://nichcy.org/disability) ii. Autism Spectrum Disorders Fact Sheet iii. Blindness/Visual Impairment Fact Sheet iv. Cerebral Palsy Fact Sheet v. Deaf-Blindness Fact Sheet vi. Deafness and Hearing Loss Fact Sheet vii. Developmental Delay Fact Sheet viii. Down Syndrome Fact Sheet ix. Emotional Disturbance Fact Sheet x. Epilepsy Fact Sheet xi. Intellectual Disabilities Fact Sheet xii. Learning Disabilities Fact Sheet xiii. Other Health Impairment Fact Sheet xiv. Traumatic Brain Injury Fact Sheet b. Disability Worksheets for Eligibility for Special Education (from OSSE/DCPS) i. Other Health Impairment Disability Worksheet ii. Specific Learning Disability Worksheet iii. Emotional Disturbance Disability Worksheet Attention-Deficit/ Hyperactivity Disorder NICHCY Disability Fact Sheet #19 Updated March 2012 break down his lessons into gets to choose something fun several parts. Then they have he’d like to do. Having a him do each part one at a child with AD/HD is still a Mario’s Story time. This helps Mario keep challenge, but things are his attention on his work. looking better. Mario is 10 years old. When he was 7, his family At home, things have learned he had AD/HD. At changed, too. Now his What is AD/HD? the time, he was driving parents know why he’s so everyone crazy. At school, he active. They are careful to Attention-deficit/hyperac- couldn’t stay in his seat or praise him when he does tivity disorder (AD/HD) is a keep quiet. At home, he something well. -

Genetic Testing: Background and Policy Issues
Genetic Testing: Background and Policy Issues Amanda K. Sarata Specialist in Health Policy March 2, 2015 Congressional Research Service 7-5700 www.crs.gov RL33832 Genetic Testing: Background and Policy Issues Summary Congress has considered, at various points in time, numerous pieces of legislation that relate to genetic and genomic technology and testing. These include bills addressing genetic discrimination in health insurance and employment; precision medicine; the patenting of genetic material; and the oversight of clinical laboratory tests (in vitro diagnostics), including genetic tests. The focus on these issues signals the growing importance of public policy issues surrounding the clinical and public health implications of new genetic technology. As genetic technologies proliferate and are increasingly used to guide clinical treatment, these public policy issues are likely to continue to garner attention. Understanding the basic scientific concepts underlying genetics and genetic testing may help facilitate the development of more effective public policy in this area. Humans have 23 pairs of chromosomes in the nucleus of most cells in their bodies. Chromosomes are composed of deoxyribonucleic acid (DNA) and protein. DNA is composed of complex chemical substances called bases. Proteins are fundamental components of all living cells, and include enzymes, structural elements, and hormones. A gene is the section of DNA that contains the sequence which corresponds to a specific protein. Though most of the genome is similar between individuals, there can be significant variation in physical appearance or function between individuals due to variations in DNA sequence that may manifest as changes in the protein, which affect the protein’s function. -

Speech/Language Impairment Sheet
West Virginia State Department of Education Office of Special Education * 1-800-558-2696 * http://wvde.state.wv.us/osp/ Fact Speech/Language Impairment Sheet DEFINITION Voice: Speech impairment where the child’s voice has an IDEA defines a speech or language impairment as a abnormal quality of pitch, resonance, or loudness. communication disorder, such as stuttering, impaired articulation, a language impairment, or a voice Persistent voice issues can indicate problems such as impairment that adversely affects a child’s educational vocal cord nodules, which should be addressed. Some performance. students who have structural abnormalities, such as cleft palate or palatal insufficiency, may exhibit a nasal SPEECH DISORDERS sound to their speech. An otolaryngologist must verify Speech Sound Disorders: Speech impairment where the the absence or existence of structural or functional child has difficulty producing sounds. Most children pathology before voice therapy can be provided. make some mistakes as they learn to say new words. A speech sound disorder occurs when mistakes continue Dysphagia: Some children may have difficulty eating past a certain age. Every sound has a different range of (chewing and swallowing food) and drinking without ages when the child should make the sound correctly. aspiration. The speech-language pathologist may be involved in developing a plan, along with other team Speech sound disorders include problems with members, to ensure that the child is provided with safe articulation (making sounds) and phonological nourishment and hydration during school hours. processes (patterns of sounds). COMMUNICATION DISORDERS Sounds can be substituted (saying “tat” for “cat”), left Augmentative and Alternative Communication: the use off (saying “ca” instead of “cat”), added (saying “kwat” of an alternative to speaking as a substitute for speech instead of “cat”) or changed (saying the sound or to supplement speech. -

Early Help Better Future
Early Help Better Future A Guide to the Early Recognition of Dyslexia by Jean Augur Introduction In the past it was thought that the earliest that a child could be identified as having a dyslexic profile was at about the age of six. This was because, by six, the child was already giving cause for concern particularly as regards reading, writing and spelling, all very important skills in the school curriculum. With experience, however, and from the findings of research studies, it is now evident that there are many signs well before school age which may suggest such a profile and the consequent difficulties ahead. Parents and pre-school carers as well as educators in those early years are amongst those in the best position to recognise these signs, and to provide appropriate activities to help. Training in some of these activities will help to build firm foundations for later, more formal, training. What is Dyslexia? Dyslexia is best described as a specific difficulty in learning, in one or more of reading, spelling and written language which may be accompanied by difficulty in number work, short-term memory, sequencing, auditory and/or visual perception, and motor skills. It is particularly related to mastering and using written language – alphabetic, numeric and musical notation. In addition, oral language is often affected to some degree. Dyslexia occurs despite normal teaching and is independent of socio-economic background or intelligence. It is, however, more easily detected in those with average or above intelligence. 1 Some of the Early Signs which may suggest a Dyslexic Profile General · Family history of similar difficulties; · May have walked early but did not crawl – was a “bottom shuffler” or “tummy wriggler”; · Persistent difficulties in getting dressed efficiently; · Persistent difficulty in putting shoes on the correct feet; Just marking the shoes with “outside” may help to ease the difficulty. -

Characterization of Point Mutations in the Same Arginine Codon in Three Unrelated Patients with Ornithine Transcarbamylase Deficiency
Characterization of point mutations in the same arginine codon in three unrelated patients with ornithine transcarbamylase deficiency. A Maddalena, … , W E O'Brien, R L Nussbaum J Clin Invest. 1988;82(4):1353-1358. https://doi.org/10.1172/JCI113738. Research Article Point mutations in the X-linked ornithine transcarbamylase (OTC) gene have been detected at the same Taq I restriction site in 3 of 24 unrelated probands with OTC deficiency. A de novo mutation could be traced in all three families to an individual in a prior generation, confirming independent recurrence. The DNA sequence in the region of the altered Taq I site was determined in the three probands. In two unrelated male probands with neonatal onset of severe OTC deficiency, a guanine (G) to adenine (A) mutation on the sense strand (antisense cytosine [C] to thymine [T]) was found, resulting in glutamine for arginine at amino acid 109 of the mature polypeptide. In the third case, where the proband was a symptomatic female, C to T (sense strand) transition converted residue 109 to a premature stop. These results support the observation that Taq I restriction sites, which contain an internal CG, are particularly susceptible to C to T transition mutation due to deamination of a methylated C in either the sense or antisense strand. The OTC gene seems especially sensitive to C to T transition mutation at arginine codon 109 because either a nonsense mutation or an extremely deleterious missense mutation will result. Find the latest version: https://jci.me/113738/pdf Characterization of Point Mutations in the Same Arginine Codon in Three Unrelated Patients with Ornithine Transcarbamylase Deficiency Anne Maddalena,*" J. -

Indel Information Eliminates Trivial Sequence Alignment in Maximum Likelihood Phylogenetic Analysis
Cladistics Cladistics 28 (2012) 514–528 10.1111/j.1096-0031.2012.00402.x Indel information eliminates trivial sequence alignment in maximum likelihood phylogenetic analysis John S.S. Dentona,b,* and Ward C. Wheelerb,c aDivision of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA; bRichard Gilder Graduate School, American Museum of Natural History, New York, NY 10024, USA; cDivision of Invertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA Accepted 12 March 2012 Abstract Although there has been a recent proliferation in maximum-likelihood (ML)-based tree estimation methods based on a fixed sequence alignment (MSA), little research has been done on incorporating indel information in this traditional framework. We show, using a simple model on a single character example, that a trivial alignment of a different form than that previously identified for parsimony is optimal in ML under standard assumptions treating indels as ‘‘missing’’ data, but that it is not optimal when indels are incorporated into the character alphabet. We show that the optimality of the trivial alignment is not an artefact of simplified theory assumptions by demonstrating that trivial alignment likelihoods of five different multiple sequence alignment datasets exhibit this phenomenon. These results demonstrate the need for use of indel information in likelihood analysis on fixed MSAs, and suggest that caution must be exercised when drawing conclusions from software implementations claiming improvements in likelihood scores under an indels-as-missing assumption. Ó The Willi Hennig Society 2012. Maximum likelihood (ML) has become a popular the increasing sophistication of search heuristics in ML optimality criterion for inferring evolutionary trees software (e.g. -
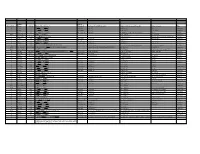
Systematic (Genomic Based)
Trivial Codon Exon/Intron Name Codon Nucleotide Change* Changed Systematic(cDNA based) Systematic (genomic based) Other names Type 2 M-1T -1 CCATGGC>CCACGGC Met>Thr c.2T>C g.4895T>C M1T transition 2 R3Op 3 CACCGAT>CACTGAT Arg>Opal c.10C>T g.4903C>T R3X, R3ter transition 2 ∆A20 20 CCC[A]GAG>CCCGAG Gln>Arg (fs) c.62delA g.4955delA (first base of deletion) ∆A20 deletion IVS2 IVS2-1G>A atagGTA>ataaGTA g.5814G>A c.113-1G>A transition 3 IVS2-1∆4 37-38 ata[gGTA]CCA>ataCCA g.5813delGGTA G12/∆3;IVS2-delGGTA;∆4IVS2-E3 deletion 3 R59op 59 TTCCGAG>TTCTGAG Arg>Opal c.178C>T g.5880C>T R59X;R59ter transition 3 I73T 73 GCATCGG>GCACCGG Ile>Thr c.221T>C g.5923T>C transition 3 L83∆C 83 CCT[C]TAC>CCTTAC Leu>Leu(fs) c.252delC g.5954delC c.250 delC deletion 3 1123ins12 104 GGT[]GGG>GGT[GGGGATCGTGGT]GGG c.314^315ins12 g.6016^6017insGGGGATCGTGGT —12E3;V104+GIVV insertion IVS3 K107K∆12 104-107del GTG[GTGGGAATCAAG]gtt>GTGgtgggaatcaaagtt c.324G>A;delGTGGGAATCAAG(312-324) g.6026G>A g.1133G>A transition IVS3 IVS3-1G>A acagTTA>acaaTTA g.7257G>A c.325-1G>C, g.8180G>C,IVS3sas transversion 4 ∆28E4 114-151 TCC[TCTTGCAGGAACAAACAAAGAAACCACC]ATT>TCCATT Pro>Pro(fs) c.345-372del28 g.7277^7305del c.345_72del28 deletion 4 Q110Oc 110 GACCAAG>GACTAAG Gln>Ochre c.331C>T g.7264C>T transition 4 ∆4E4 118-120 GAAC[AAAC]AAAG>GAACAAAG c.357delAAAC g.7289delAAAC ∆4, MD∆4 deletion 4-5 ∆E4-E5 ccc[tgt…TCC]AGC>cccAGC g.6594^8239del F13/∆4,5 deletion 5 C134R 134 CGCTGTG>CGCCGTG Cys>Arg c.403T>C g.8162T>C C135R transition 5 W147R 147 AAGTGGC>AAGCGGC Trp>Arg c.442T>C g.8201T>C W148R transition -

Part 7 Developmental Disabilities
Bringing It All Together You are a group leader on one of the ICF units at SDC, one of the clients in your group is Mickey, a 44-year-old man with Down Syndrome. He is sociable, but mischievous. He can say a few words, such as "toy", "no", and "hurt." He enjoys his vocational activity most of the time but has to be reminded, repeatedly. to get back to work (he has a job shredding paper). His physical treatment profile lists: hypothyroidism, constipation, dermatitis, GERD, and obstructive sleep apnea. When he is angry he vocalizes and pushes people; when he's happy he can be a charmer. 1. As a psychiatric technician you know that Down syndrome is: a. A disorder caused by gestational problems in the neonate b. A genetic abnormality c. Perinatal hypoxia d. Drug exposure during pregnancy Answer: B- Down syndrome, also called Trisomy 21- is a genetic disorder. Perinatal hypoxia has long been suspected of causing cerebral palsy, but there may be other factors involved. Drug exposure during pregnancy is usually worst with resultant F AS. 2. The common characteristics of Down are: a. Short stature, epicanthal fold, smallish head b. Long face, hand flapping c. .:Ylicrocephaly, hirshutism, and severe mental impairment d. Port wine stain Answer: A-Down syndrome. A more complete list would include: Poor muscle tone; Slanting eyes with folds of skin at the inner corners (callcd epicanthal folds); Hyperflexibility (excessive ability to extend the joints); Short, broad hands with a single crease across the palm on one or both hands; Broad feet with short toes; Flat bridge of the nose; Short, low-set ears; Short neck; Small head; Small oral cavity; and/or short, high-pitched cries in infancy. -

G14TBS Part II: Population Genetics
G14TBS Part II: Population Genetics Dr Richard Wilkinson Room C26, Mathematical Sciences Building Please email corrections and suggestions to [email protected] Spring 2015 2 Preliminaries These notes form part of the lecture notes for the mod- ule G14TBS. This section is on Population Genetics, and contains 14 lectures worth of material. During this section will be doing some problem-based learning, where the em- phasis is on you to work with your classmates to generate your own notes. I will be on hand at all times to answer questions and guide the discussions. Reading List: There are several good introductory books on population genetics, although I found their level of math- ematical sophistication either too low or too high for the purposes of this module. The following are the sources I used to put together these lecture notes: • Gillespie, J. H., Population genetics: a concise guide. John Hopkins University Press, Baltimore and Lon- don, 2004. • Hartl, D. L., A primer of population genetics, 2nd edition. Sinauer Associates, Inc., Publishers, 1988. • Ewens, W. J., Mathematical population genetics: (I) Theoretical Introduction. Springer, 2000. • Holsinger, K. E., Lecture notes in population genet- ics. University of Connecticut, 2001-2010. Available online at http://darwin.eeb.uconn.edu/eeb348/lecturenotes/notes.html. • Tavar´eS., Ancestral inference in population genetics. In: Lectures on Probability Theory and Statistics. Ecole d'Et´esde Probabilit´ede Saint-Flour XXXI { 2001. (Ed. Picard J.) Lecture Notes in Mathematics, Springer Verlag, 1837, 1-188, 2004. Available online at http://www.cmb.usc.edu/people/stavare/STpapers-pdf/T04.pdf Gillespie, Hartl and Holsinger are written for non-mathematicians and are easiest to follow. -

A Review of Tools/Objects Used in Articulation Therapy by Van Riper and Other Traditional Therapists
Volume 37 Number 1 pp. 69-96 2011 Review Article Horns, whistles, bite blocks, and straws: A review of tools/objects used in articulation therapy by van Riper and other traditional therapists Pam Marshalla (Marshalla Speech and Language) Follow this and additional works at: https://ijom.iaom.com/journal The journal in which this article appears is hosted on Digital Commons, an Elsevier platform. Suggested Citation Marshalla, P. (2011). Horns, whistles, bite blocks, and straws: A review of tools/objects used in articulation therapy by van Riper and other traditional therapists. International Journal of Orofacial Myology, 37(1), 69-96. DOI: https://doi.org/10.52010/ijom.2011.37.1.6 This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. The views expressed in this article are those of the authors and do not necessarily reflect the policies or positions of the International Association of Orofacial Myology (IAOM). Identification of specific oducts,pr programs, or equipment does not constitute or imply endorsement by the authors or the IAOM. International Journal of Orofacial Myology 2011, V37 HORNS, WHISTLES, BITE BLOCKS, AND STRAWS: A REVIEW OF TOOLS/OBJECTS USED IN ARTICULATION THERAPY BY VAN RIPER AND OTHER TRADITIONAL THERAPISTS PAM MARSHALLA, MA, CCC-SLP ABSTRACT The use of tools and other objects in articulation therapy has been bundled into new groups of activities called “nonspeech oral motor exercises” (NSOME) and ‘nonspeech oral motor treatments’ (NSOMT) by some authors. The purveyors of these new terms suggest that there is no proof that such objects aid speech learning, and they have cautioned students and professionals about their use. -

CRISPR-Cas9 DNA Base-Editing and Prime-Editing
International Journal of Molecular Sciences Review CRISPR-Cas9 DNA Base-Editing and Prime-Editing Ariel Kantor 1,2,*, Michelle E. McClements 1,2 and Robert E. MacLaren 1,2 1 Nuffield Laboratory of Ophthalmology, Nuffield Department of Clinical Neurosciences & NIHR Oxford Biomedical Research Centre, University of Oxford, Oxford OX3 9DU, UK 2 Oxford Eye Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford OX3 9DU, UK * Correspondence: [email protected] Received: 14 July 2020; Accepted: 25 August 2020; Published: 28 August 2020 Abstract: Many genetic diseases and undesirable traits are due to base-pair alterations in genomic DNA. Base-editing, the newest evolution of clustered regularly interspaced short palindromic repeats (CRISPR)-Cas-based technologies, can directly install point-mutations in cellular DNA without inducing a double-strand DNA break (DSB). Two classes of DNA base-editors have been described thus far, cytosine base-editors (CBEs) and adenine base-editors (ABEs). Recently, prime-editing (PE) has further expanded the CRISPR-base-edit toolkit to all twelve possible transition and transversion mutations, as well as small insertion or deletion mutations. Safe and efficient delivery of editing systems to target cells is one of the most paramount and challenging components for the therapeutic success of BEs. Due to its broad tropism, well-studied serotypes, and reduced immunogenicity, adeno-associated vector (AAV) has emerged as the leading platform for viral delivery of genome editing agents, including DNA-base-editors. In this review, we describe the development of various base-editors, assess their technical advantages and limitations, and discuss their therapeutic potential to treat debilitating human diseases.