Rapidly Progressive Dementia (RPD)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Encephalopathy and Encephalitis Associated with Cerebrospinal
SYNOPSIS Encephalopathy and Encephalitis Associated with Cerebrospinal Fluid Cytokine Alterations and Coronavirus Disease, Atlanta, Georgia, USA, 2020 Karima Benameur,1 Ankita Agarwal,1 Sara C. Auld, Matthew P. Butters, Andrew S. Webster, Tugba Ozturk, J. Christina Howell, Leda C. Bassit, Alvaro Velasquez, Raymond F. Schinazi, Mark E. Mullins, William T. Hu There are few detailed investigations of neurologic unnecessary staff exposure and difficulties in estab- complications in severe acute respiratory syndrome lishing preillness neurologic status without regular coronavirus 2 infection. We describe 3 patients with family visitors. It is known that neurons and glia ex- laboratory-confirmed coronavirus disease who had en- press the putative SARS-CoV-2 receptor angiotensin cephalopathy and encephalitis develop. Neuroimaging converting enzyme 2 (7), and that the related coro- showed nonenhancing unilateral, bilateral, and midline navirus SARS-CoV (responsible for the 2003 SARS changes not readily attributable to vascular causes. All 3 outbreak) can inoculate the mouse olfactory bulb (8). patients had increased cerebrospinal fluid (CSF) levels If SARS-CoV-2 can enter the central nervous system of anti-S1 IgM. One patient who died also had increased (CNS) directly or through hematogenous spread, ce- levels of anti-envelope protein IgM. CSF analysis also rebrospinal fluid (CSF) changes, including viral RNA, showed markedly increased levels of interleukin (IL)-6, IgM, or cytokine levels, might support CNS infec- IL-8, and IL-10, but severe acute respiratory syndrome coronavirus 2 was not identified in any CSF sample. tion as a cause for neurologic symptoms. We report These changes provide evidence of CSF periinfectious/ clinical, blood, neuroimaging, and CSF findings for postinfectious inflammatory changes during coronavirus 3 patients with laboratory-confirmed COVID-19 and disease with neurologic complications. -

TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy
J Neuropathol Exp Neurol Vol. 69, No. 9 Copyright Ó 2010 by the American Association of Neuropathologists, Inc. September 2010 pp. 918Y929 ORIGINAL ARTICLE TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy Ann C. McKee, MD, Brandon E. Gavett, PhD, Robert A. Stern, PhD, Christopher J. Nowinski, AB, Robert C. Cantu, MD, Neil W. Kowall, MD, Daniel P. Perl, MD, E. Tessa Hedley-Whyte, MD, Bruce Price, MD, Chris Sullivan, Peter Morin, MD, PhD, Hyo-Soon Lee, MD, Caroline A. Kubilus, Daniel H. Daneshvar, MA, Megan Wulff, MPH, and Andrew E. Budson, MD cord in addition to tau neurofibrillary changes, motor neuron loss, Abstract and corticospinal tract degeneration. The TDP-43 proteinopathy Epidemiological evidence suggests that the incidence of amyo- associated with CTE is similar to that found in frontotemporal lobar trophic lateral sclerosis is increased in association with head injury. degeneration with TDP-43 inclusions, in that widespread regions of Repetitive head injury is also associated with the development of the brain are affected. Akin to frontotemporal lobar degeneration chronic traumatic encephalopathy (CTE), a tauopathy characterized with TDP-43 inclusions, in some individuals with CTE, the TDP-43 by neurofibrillary tangles throughout the brain in the relative absence proteinopathy extends to involve the spinal cord and is associated of A-amyloid deposits. We examined 12 cases of CTE and, in 10, with motor neuron disease. This is the first pathological evidence that found a widespread TAR DNA-binding protein of approximately repetitive head trauma experienced in collision sports might be 43 kd (TDP-43) proteinopathy affecting the frontal and temporal associated with the development of a motor neuron disease. -

Encephalopathy
Jounal ofNeurology, Neurosurgery, and Psychiatry 1997;62:51-60 51 Operational criteria for the classification of chronic alcoholics: identification of Wemicke's encephalopathy D Caine, G M Halliday, J J Kril, C G Harper Abstract suggest that there can be a complete resolution Objectives-To establish better opera- of the underlying brain abnormality in tional criteria for the diagnosis of Wernicke's encephalopathy, similar to the res- Wernicke's encephalopathy. Current olution of neuropathology in hepatic criteria for diagnosing Wernicke's encephalopathy. However, unlike hepatic encephalopathy require the presence of encephalopathy, in which the clinical signs are three clinical signs (oculomotor abnor- a precursor of the pathology, many patients malities, cerebellar dysfunction, and an with the neuropathology of Wernicke's altered mental state), although it has encephalopathy do not have recorded signs of often been reported that most patients do the classic triad.' 2 This raises the question of not filfil all these criteria. whether the clinical correlates of the patholog- Methods-The clinical histories of 28 ical lesions are being missed during life or alcoholics with neurological and neu- whether the pathology represents clinically ropsychological assessments and defini- silent lesions. A similar situation is seen in tive neuropathological diagnoses were Alzheimer's disease, in which many non- examined to determine clinical signs for demented aged patients show a similar type use in a screening schedule. Operational and distribution of neuropathology to their criteria were then proposed for differenti- demented counterparts. 12 Recently research ating patients with Wernicke's into the pathophysiology of aging and encephalopathy alone or in combination Alzheimer's disease has been successfully with Korsakoff's psychosis or hepatic advanced by the use of operational criteria encephalopathy. -

Pharmacologic Treatment of Meningitis and Encephalitis in Adult Patients
Published online: 2019-06-19 THIEME Review Article 145 Pharmacologic Treatment of Meningitis and Encephalitis in Adult Patients Andrew K. Treister1 Ines P. Koerner2 1Department of Neurology, Oregon Health & Science University, Address for correspondence Andrew K. Treister, MD, Department Portland, Oregon, United States of Neurology, Oregon Health & Science University, 3181 SW Sam 2Department of Anesthesiology and Perioperative Medicine, Jackson Park, CR 120, Portland, OR 97239-3098, United States Oregon Health & Science University, Portland, Oregon, (e-mail: [email protected]). United States J Neuroanaesthesiol Crit Care 2019;6:145–152 Abstract Meningitis and encephalitis are two inflammatory, often infectious, disorders of the meninges and the central nervous system. Both are associated with significant morbidity and mortality, and require early and aggressive targeted treatment. This article reviews pharmacologic treatment strategies for infectious meningitis and encephalitis, using Keywords the latest available research and guidelines. It provides an overview of empiric anti- ► meningitis microbial treatment approaches for a variety of organisms, including a discussion of ► encephalitis trends in antibiotic resistance where applicable. Key steps in diagnosis and general ► antibiotic resistance management are briefly reviewed. Introduction care–associated infections are not covered in detail, as they are excellently discussed in a recent guideline statement.4 The terms meningitis and encephalitis comprise a broad array of infectious and inflammatory processes involving the central nervous system (CNS) that carry significant morbidity Literature Review 1,2 and mortality. Meningitis refers to inflammation of PubMed was searched in January 2019 for articles published primarily the meninges, although it may spread to involve the between January 1, 2009, and December 31, 2018. -

Idiopathic Intracranial Hypertension
IDIOPATHIC INTRACRANIAL HYPERTENSION William L Hills, MD Neuro-ophthalmology Oregon Neurology Associates Affiliated Assistant Professor Ophthalmology and Neurology Casey Eye Institute, OHSU No disclosures CASE - 19 YO WOMAN WITH HEADACHES X 3 MONTHS Headaches frontal PMHx: obesity Worse lying down Meds: takes ibuprofen for headaches Wake from sleep Pulsatile tinnitus x 1 month. Vision blacks out transiently when she bends over or sits down EXAMINATION Vision: 20/20 R eye, 20/25 L eye. Neuro: PERRL, no APD, EOMI, VF full to confrontation. Dilated fundoscopic exam: 360 degree blurring of disc margins in both eyes, absent SVP. Formal visual field testing: Enlargement of the blind spot, generalized constriction both eyes. MRI brain: Lumbar puncture: Posterior flattening of Opening pressure 39 the globes cm H20 Empty sella Normal CSF studies otherwise normal Headache improved after LP IDIOPATHIC INTRACRANIAL HYPERTENSION SYNDROME: Increased intracranial pressure without ventriculomegaly or mass lesion Normal CSF composition NOMENCLATURE Idiopathic intracranial hypertension (IIH) Benign intracranial hypertension Pseudotumor cerebri Intracranial hypertension secondary to… DIAGNOSTIC CRITERIA Original criteria have been updated to reflect new imaging modalities: 1492 Friedman and Jacobsen. Neurology 2002; 59: Symptoms and signs reflect only those of - increased ICP or papilledema 1495 Documented increased ICP during LP in lateral decubitus position Normal CSF composition No evidence of mass, hydrocephalus, structural -
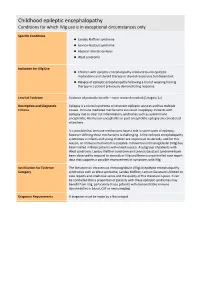
Childhood Epileptic Encephalopathy Conditions for Which Ivig Use Is in Exceptional Circumstances Only
Childhood epileptic encephalopathy Conditions for which IVIg use is in exceptional circumstances only Specific Conditions Landau Kleffner syndrome Lennox‐Gastaut syndrome Atypical rolandic epilepsy West syndrome Indication for IVIg Use Children with epileptic encephalopathy resistant to anti‐epileptic medications and steroid therapy or steroid responsive but dependant Relapse of epileptic encephalopathy following a trial of weaning from Ig therapy in a patient previously demonstrating response Level of Evidence Evidence of probable benefit – more research needed (Category 2a) Description and Diagnostic Epilepsy is a clinical syndrome of recurrent epileptic seizures and has multiple Criteria causes. Immune mediated mechanisms can result in epilepsy. Patients with epilepsy due to clear cut inflammatory syndromes such as autoimmune encephalitis, Rasmussen encephalitis or post encephalitic epilepsy are considered elsewhere. It is possible that immune mechanisms have a role in some cases of epilepsy, however defining these mechanisms is challenging. A few epileptic encephalopathy syndromes in infants and young children are responsive to steroids, and for this reason, an immune mechanism is possible. Intravenous immunoglobulin (IVIg) has been trialled in these patients with mixed success. A subgroup of patients with West syndrome, Landau Kleffner syndrome and Lennox Gaustaut syndrome have been observed to respond to steroids or IVIg and there is uncontrolled case report data that supports a possible improvement of symptoms with IVIg. Justification for Evidence The literature on intravenous immunoglobulin (IVIg) in epileptic encephalopathy Category syndromes such as West syndrome, Landau Kleffner, Lennox Gaustaut is limited to case reports and small case series and the quality of this literature is poor. It can be concluded that a proportion of patients with these epileptic syndromes may benefit from IVIg, particularly those patients with demonstrable immune abnormalities in blood, CSF or neuroimaging. -

Encephalopathy
Causes Encephalopathy (Dementia) This condition is caused when: Encephalopathy is the name of several brain HIV blocks how your brain cells talk to each other problems linked to AIDS that cause memory and how they talk to the rest of your body problems. These problems can be very mild or severe. There are many names for these How to Know You Have Encephalopathy problems. Some are worse than others. You will need a doctor’s exam to know you have • HIV encephalopathy this illness. • HIV-associated progressive encephalopathy in He or she may order: children • A mental status exam. This checks how well a • HIV-associated dementia person can recall things, focus, move, learn, talk • HIV mild neurocognitive disorder and use words and ideas. • Asymptomatic neurocognitive impairment • A spinal tap. This takes fluid from around the brain and spinal cord to make sure you don’t HIV meds make the severe problems much less have an infection other than HIV. common. • Blood tests to make sure you don’t have Signs syphilis, thyroid disease or low b12 This disease can cause: • Tests that look at your brain, such as a CT or • Loss of interest in life, other people, hobbies or MRI scan sex. A boredom with life. • A doctor to help manage depression • Memory problems • A visit to a brain doctor • Lack of focus Treatment • Forget things like taking your meds HIV meds are the best way to treat this. • Problems on your job Sometimes your doctor may give you meds that • Loss of bladder control get into the brain better. -

Clinical Documentation Improvement Faqs of BLR AACDIFAQ
ACDIS Answers Clinical Documentation FAQs | Clinical Documentation Improvement Answers ACDIS Improvement FAQs This compendium of commonly asked CDI questions is an essential reference book and office companion, valuable for new CDI specialists as well as those experienced in concurrent medical record review. Whether you’re wondering about ACDIS Answers sequencing guidelines, staff productivity, escalation policies, diabetes coding, or documentation requirements for acute kidney injury, ACDIS Answers provides quick, easily understandable Clinical Documentation information from respected experts in CDI, including ACDIS’ own Boot Camp instructors and Advisory Board members. Improvement FAQs AACDIFAQ of BL a division 100 Winners Circle, Suite 300 R Brentwood, TN 37027 www.hcmarketplace.com 35690_EB334325_AACDIFAQ_Cover.indd 1 12/13/16 10:55 AM ACDIS Answers: Clinical Documentation Improvement FAQs LAURIE L. PRESCOTT, MSN, RN, CCDS, CDIP, CRC SHARME BRODIE, RN, CCDS ALLEN FRADY, RN, BSN, CCDS, CCS ACDIS Answers: Clinical Documentation Improvement FAQsis published by HCPro, a division of BLR. Copyright © 2016 HCPro, a division of BLR All rights reserved. Printed in the United States of America. 5 4 3 2 1 ISBN: 978-1-68308-207-1 No part of this publication may be reproduced, in any form or by any means, without prior written consent of HCPro or the Copyright Clearance Center (978-750-8400). Please notify us immediately if you have received an unauthorized copy. HCPro provides information resources for the healthcare industry. HCPro is not affiliated in any way with The Joint Commission, which owns the JCAHO and Joint Commission trademarks. Laurie L. Prescott, MSN, RN, CCDS, CDIP, CRC, Author Sharme Brodie, RN, CCDS, Author Allen Frady, RN, BSN, CCDS, CCS, Author Katherine Rushlau, Editor Melissa Varnavas, Editor Erin Callahan, Vice President, Product Development & Content Strategy Elizabeth Petersen, Executive Vice President, Healthcare Matt Sharpe, Production Supervisor Vincent Skyers, Design Services Director Vicki McMahan, Sr. -

ADEM) After Autologous Peripheral Blood Stem Cell Transplant for Non-Hodgkin’S Lymphoma
Bone Marrow Transplantation, (1999) 24, 1351–1354 1999 Stockton Press All rights reserved 0268–3369/99 $15.00 http://www.stockton-press.co.uk/bmt Case report Acute disseminated encephalomyelitis (ADEM) after autologous peripheral blood stem cell transplant for non-Hodgkin’s lymphoma A Re and R Giachetti Department of Hematology, University of Parma, Italy Summary: High-dose chemotherapy followed by autologous periph- eral blood stem cell transplantation (PBSCT) is a thera- Acute disseminated encephalomyelitis (ADEM) is a peutic intervention performed with increasing frequency for demyelinating disorder of the central nervous system hematologic and solid malignancies.4 The widespread use with an acute clinical onset and a wide variability in of this procedure depends on its safety and easy feasibility. severity and outcome. It usually follows a viral infection Mild organ toxicities and low incidence of life-threatening or an immunization and is thought to be immuno- complications are usually reported. Neurologic events are mediated. We report a case of ADEM with a dramatic frequently mild and reversible and usually secondary to clinical onset in an autologous peripheral blood stem injury to other organ systems.5 cell transplant (PBSCT) recipient for non-Hodgkin’s We report a case of ADEM in an autologous PBSCT lymphoma who developed the neurologic syndrome 12 recipient with non-Hodgkin’s lymphoma (NHL) who days after PBSC reinfusion. This is the first report of developed the syndrome on day ϩ12 after PBSC ADEM in the setting of autologous PBSCT, a thera- reinfusion, without any recognizable etiology. To our peutic procedure performed with increasing frequency knowledge, this is the first report of ADEM developing in a wide variety of hematologic and solid malignancies. -

Toxic Encephalopathy
Toxic Encephalopathy Jacob Valk 1 and f\1. S. van der Knaap From the Departments of Diagnostic Radiology (JV) and Child Neurology (MSvdK), the Free University Hospital, Amsterdam, The Netherlands There is growing awareness that chronic intox 1941-Lathyrus sativus peas, spastic ications by industrial, agricultural, iatrogenic, and paraparesis; the toxic agent was environmental pollution may have teratogenic or identified to be 1)-JY-methylamino-L oncogenic influence or may cause neurologic or alanine (BMAA); psychiatric syndromes. 1953-Guamalian type of Parkinsonism, Toxic encephalopathy (TE) is the result of the caused by the seeds of Cycas interaction of a chemical compound with the circinalis; the toxic agent was brain. Disturbance of normal brain function is identified as 1)-JY-oxalylmethylamino-L caused by: alanine (BOMAA); 1. depletion of oxidative energy; 1948-hexachloraphene encephalopathy; 2. nutritional deprivation affecting nerves and 1950-monosodium glutamate in baby food; neurons; 1953-Minamata disease, mercury 3. exposure to foreign material which may be encephalopathy; a. exogenous in origin, 1960-housepainters dementia, organic b. generated within the central nervous solvents; system, or 1983-methylphenyltetrahydropyridine c. generated within the body; (MPTP), "synthetic heroin," causing 4. derangement of neurotransmission; striatal dopamine deficiency and 5. altered ion balance; Parkinsonism. 6. antigenic activity. Clinically toxic encephalopathy presents with The list of examples of toxic encephalopathy one of more of the following neurologic or psy is long and reflects the real difficulty in recogniz chiatric symptoms: ing that slow deterioration of neurologic functions 1. decreased concentration and indicates poisoning by a toxin. In many cases, consciousness; religious, superstitious, or racial "explanations" 2. -

Mechanisms, Diagnosis and Management of Hepatic Encephalopathy Ravi Prakash and Kevin D
REVIEWS Mechanisms, diagnosis and management of hepatic encephalopathy Ravi Prakash and Kevin D. Mullen Abstract | Hepatic encephalopathy (HE) is a serious neuropsychiatric complication of both acute and chronic liver disease. Symptoms of HE can include confusion, disorientation and poor coordination. A general consensus exists that the synergistic effects of excess ammonia and inflammation cause astrocyte swelling and cerebral edema; however, the precise molecular mechanisms that lead to these morphological changes in the brain are unclear. Cerebral edema occurs to some degree in all patients with HE, regardless of its grade, and could underlie the pathogenesis of this disorder. The different grades of HE can be diagnosed by a number of investigations, including neuropsychometric tests (such as the psychometric hepatic encephalopathy score), brain imaging and clinical scales (such as the West Haven criteria). HE is best managed by excluding other possible causes of encephalopathy alongside identifying and the precipitating cause, and confirming the diagnosis by a positive response to empiric treatment. Empiric therapy for HE is largely based on the principle of reducing the production and absorption of ammonia in the gut through administration of pharmacological agents such as rifaximin and lactulose, which are approved by the FDA for the treatment of HE. Prakash, R. & Mullen, K. D. Nat. Rev. Gastroenterol. Hepatol. 7, 515–525 (2010); published online 10 August 2010; doi:10.1038/nrgastro.2010.116 Introduction Continuing Medical Education online Hepatic encephalopathy (HE) is a serious neuro This activity has been planned and implemented in accordance psychiatric complication of both acute and chronic with the Essential Areas and policies of the Accreditation Council liver disease.1 This disease encompasses a broad range for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Nature Publishing Group. -

Vaccine Injury Table
Vaccine Injury Table Applies Only to Petitions for Compensation Filed under the National Vaccine Injury Compensation Program on or after March 21, 2017 (a) In accordance with section 312(b) of the National Childhood Vaccine Injury Act of 1986, title III of Public Law 99-660, 100 Stat. 3779 (42 U.S.C. 300aa-1 note) and section 2114(c) of the Public Health Service Act, as amended (PHS Act) (42 U.S.C. 300aa-14(c)), the following is a table of vaccines, the injuries, disabilities, illnesses, conditions, and deaths resulting from the administration of such vaccines, and the time period in which the first symptom or manifestation of onset or of the significant aggravation of such injuries, disabilities, illnesses, conditions, and deaths is to occur after vaccine administration for purposes of receiving compensation under the Program. Paragraph (b) of this section sets forth additional provisions that are not separately listed in this Table but that constitute part of it. Paragraph (c) of this section sets forth the qualifications and aids to interpretation for the terms used in the Table. Conditions and injuries that do not meet the terms of the qualifications and aids to interpretation are not within the Table. Paragraph (d) of this section sets forth a glossary of terms used in paragraph (c). Time period for first symptom or manifestation of onset or of Illness, disability, injury significant aggravation after Vaccine or condition covered vaccine administration I. Vaccines containing tetanus toxoid (e.g., A. Anaphylaxis ≤4 hours. DTaP, DTP, DT, Td, or TT) B. Brachial Neuritis 2-28 days (not less than 2 days and not more than 28 days).