Clinical and Molecular Characterization of Achromatopsia Patients: a Longitudinal Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Masqueraders of Age-Related Macular Degeneration
COVER STORY Masqueraders of Age-related Macular Degeneration A number of inherited retinal diseases phenocopy AMD. BY RONY GELMAN, MD, MS; AND STEPHEN H. TSANG, MD, PHD ge-related macular degeneration (AMD) is a leading cause of central visual loss among the elderly population in the developed world. The Currently, there are no published A non-neovascular form is characterized by mac- guidelines to prognosticate ular drusen and other abnormalities of the retinal pigment epithelium (RPE) such as geographic atrophy (GA) and Stargardt macular degeneration. hyperpigmented areas in the macula. The neovascular form is heralded by choroidal neovascularization (CNV), with subsequent development of disciform scarring. ABCA4 defect heterozygote carrier may be as high as This article reviews the pathologic and diagnostic char- one in 20.11,12 An estimated 600 disease-causing muta- acteristics of inherited diseases that may masquerade as tions in the ABCA4 gene exist, of which the three most AMD. The review is organized by the following patterns common mutations account for less than 10% of the of inheritance: autosomal recessive (Stargardt disease and disease phenotypes.13 cone dystrophy); autosomal dominant (cone dystrophy, The underlying pathology of disease in STGD involves adult vitelliform dystrophy, pattern dystrophy, North accumulation of lipofuscin in the RPE through a process Carolina macular dystrophy, Doyne honeycomb dystro- of disc shedding and phagocytosis.14,15 Lipofuscin is toxic phy, and Sorsby macular dystrophy); X-linked (X-linked to the RPE; furthermore, A2E, a component of lipofuscin, retinoschisis); and mitochondrial (maternally inherited causes inhibition of 11-cis retinal regeneration16 and diabetes and deafness). complement activation. -

Ametropia and Emmetropization in CNGB3 Achromatopsia
Retina Ametropia and Emmetropization in CNGB3 Achromatopsia Mette Kjøbæk Gundestrup Andersen1 and Line Kessel1,2 1Department of Ophthalmology, Copenhagen University Hospital, Rigshospitalet-Glostrup, Glostrup, Denmark 2Department of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark Correspondence: Mette K.G. PURPOSE. Emmetropization is the process of adjusting ocular growth to the focal plane Andersen, Department of in order to achieve a clear image. Chromatic light may be involved as a cue to guide Ophthalmology, Copenhagen this process. Achromats are color blind and lack normal cone function; they are often University Hospital, described as being hyperopic, indicating a failure to emmetropize. We aim to describe Rigshospitalet-Glostrup, Valdemar the refraction and refractive development in a population of genetically characterized Hansens Vej 1-23, 2600 Glostrup, Denmark; achromats. [email protected]. METHODS. Refractive error data were collected retrospectively from 28 medical records CNGB3 Received: August 23, 2020 of c.1148delC homozygous achromats. The distribution of spherical equivalent Accepted: January 18, 2021 refractive error (SER) and spherical error was analyzed in adults. The refractive develop- Published: February 9, 2021 ment in children was analyzed by documenting astigmatic refractive error and calculating Citation: Andersen MKG, Kessel L. median SER in 1-year age groups and by analyzing the individual development when Ametropia and emmetropization in possible. CNGB3 Invest achromatopsia. RESULTS. The distribution of SER and spherical error resembled a Gaussian distribution, Ophthalmol Vis Sci. 2021;62(2):10. indicating that emmetropization was disturbed in achromats, but we found indication of https://doi.org/10.1167/iovs.62.2.10 some decrease in SER during the first years of childhood. -

Genetic Defects of CHM and Visual Acuity Outcome in 24 Choroideremia
www.nature.com/scientificreports OPEN Genetic defects of CHM and visual acuity outcome in 24 choroideremia patients from 16 Japanese families Takaaki Hayashi1,2*, Shuhei Kameya3, Kei Mizobuchi2, Daiki Kubota3, Sachiko Kikuchi3, Kazutoshi Yoshitake4, Atsushi Mizota5, Akira Murakami6, Takeshi Iwata4 & Tadashi Nakano2 Choroideremia (CHM) is an incurable progressive chorioretinal dystrophy. Little is known about the natural disease course of visual acuity in the Japanese population. We aimed to investigate the genetic spectrum of the CHM gene and visual acuity outcomes in 24 CHM patients from 16 Japanese families. We measured decimal best-corrected visual acuity (BCVA) at presentation and follow-up, converted to logMAR units for statistical analysis. Sanger and/or whole-exome sequencing were performed to identify pathogenic CHM variants/deletions. The median age at presentation was 37.0 years (range, 5–76 years). The mean follow-up interval was 8.2 years. BCVA of the better-seeing eye at presentation was signifcantly worsened with increasing age (r = 0.515, p < 0.01), with a high rate of BCVA decline in patients > 40 years old. A Kaplan–Meier survival curve suggested that a BCVA of Snellen equivalent 20/40 at follow-up remains until the ffties. Fourteen pathogenic variants, 6 of which were novel [c.49 + 5G > A, c.116 + 5G > A, p.(Gly176Glu, Glu177Ter), p.Tyr531Ter, an exon 2 deletion, and a 5.0-Mb deletion], were identifed in 15 families. No variant was found in one family only. Our BCVA outcome data are useful for predicting visual prognosis and determining the timing of intervention in Japanese patients with CHM variants. -

Retinitis Pigmentosa Precision Panel Overview Indications Clinical
Retinitis Pigmentosa Precision Panel Overview Retinitis Pigmentosa (RP) comprises a complex group of inherited dystrophies characterized by degeneration and dysfunction of the retina, affecting photoreceptor and pigment epithelial function. RP can be an isolated finding or be part of a syndrome that can be inherited in a dominant, recessive or X-linked pattern. This disease presents as progressive loss of night and peripheral vision, leading to a constricted visual field and markedly diminished vision. The clinical presentation of these findings is highly variable, some patients being affected during childhood while others are asymptomatic well into adulthood. There is an increase in mortality rate due to psychiatric comorbidities. The Igenomix Retinitis Pigmentosa Precision Panel can be used to make an accurate and directed diagnosis as well as a differential diagnosis of blindness ultimately leading to a better management and prognosis of the disease. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes involved. Indications The Igenomix Retinitis Pigmentosa Precision Panel is indicated for those patients with a clinical suspicion or diagnosis with or without the following manifestations: - Family history of RP - Night blindness - Progressive constriction of the visual field, usually peripheral - Cataracts - Sensation of sparking lights (photopsias) - Headache Clinical Utility The clinical utility of this panel is: - The genetic and molecular confirmation for an accurate clinical diagnosis of a symptomatic patient. - Early initiation of multidisciplinary treatment in the form of medical care with vitamin A and other antioxidants and surgical care for potential cataract extraction or retinal prosthesis. -
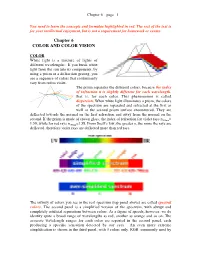
Chapter 6 COLOR and COLOR VISION
Chapter 6 – page 1 You need to learn the concepts and formulae highlighted in red. The rest of the text is for your intellectual enjoyment, but is not a requirement for homework or exams. Chapter 6 COLOR AND COLOR VISION COLOR White light is a mixture of lights of different wavelengths. If you break white light from the sun into its components, by using a prism or a diffraction grating, you see a sequence of colors that continuously vary from red to violet. The prism separates the different colors, because the index of refraction n is slightly different for each wavelength, that is, for each color. This phenomenon is called dispersion. When white light illuminates a prism, the colors of the spectrum are separated and refracted at the first as well as the second prism surface encountered. They are deflected towards the normal on the first refraction and away from the normal on the second. If the prism is made of crown glass, the index of refraction for violet rays n400nm= 1.59, while for red rays n700nm=1.58. From Snell’s law, the greater n, the more the rays are deflected, therefore violet rays are deflected more than red rays. The infinity of colors you see in the real spectrum (top panel above) are called spectral colors. The second panel is a simplified version of the spectrum, with abrupt and completely artificial separations between colors. As a figure of speech, however, we do identify quite a broad range of wavelengths as red, another as orange and so on. -

Bass – Glaucomatous-Type Field Loss Not Due to Glaucoma
Glaucoma on the Brain! Glaucomatous-Type Yes, we see lots of glaucoma Field Loss Not Due to Not every field that looks like glaucoma is due to glaucoma! Glaucoma If you misdiagnose glaucoma, you could miss other sight-threatening and life-threatening Sherry J. Bass, OD, FAAO disorders SUNY College of Optometry New York, NY Types of Glaucomatous Visual Field Defects Paracentral Defects Nasal Step Defects Arcuate and Bjerrum Defects Altitudinal Defects Peripheral Field Constriction to Tunnel Fields 1 Visual Field Defects in Very Early Glaucoma Paracentral loss Early superior/inferior temporal RNFL and rim loss: short axons Arcuate defects above or below the papillomacular bundle Arcuate field loss in the nasal field close to fixation Superotemporal notch Visual Field Defects in Early Glaucoma Nasal step More widespread RNFL loss and rim loss in the inferior or superior temporal rim tissue : longer axons Loss stops abruptly at the horizontal raphae “Step” pattern 2 Visual Field Defects in Moderate Glaucoma Arcuate scotoma- Bjerrum scotoma Focal notches in the inferior and/or superior rim tissue that reach the edge of the disc Denser field defects Follow an arcuate pattern connected to the blind spot 3 Visual Field Defects in Advanced Glaucoma End-Stage Glaucoma Dense Altitudinal Loss Progressive loss of superior or inferior rim tissue Non-Glaucomatous Etiology of End-Stage Glaucoma Paracentral Field Loss Peripheral constriction Hereditary macular Loss of temporal rim tissue diseases Temporal “islands” Stargardt’s macular due -

Retinitis Pigmentosa: a Brief Review of the Genetic and Clinical Aspects
Retinitis Pigmentosa: A Brief Review of the Genetic and Clinical Aspects of the Disease Itia Dowdell Science and Technology Honors Program, University of Alabama at Birmingham, Birmingham, AL, USA School of Health Professions Honors Program, University of Alabama at Birmingham, Birmingham, AL, USA Department of Clinical and Diagnostic Sciences, University of Alabama at Birmingham, Birmingham, AL, USA Abstract Retinitis Pigmentosa (RP) is a heterogeneous set of inherited retinal diseases that affects 1 in 3,000–7,000 people worldwide. Typical onset is from 10–30 years old and most forms are progressive, often leading to blindness. Defects in more than 200 genes have been identified that cause RP. The disease is characterized as a progressive rod-cone dystrophy that presents with night blindness, loss of peripheral vision, waxy pallor of the optic disc, pigmentary changes, and a reduced visual field. There are different modes of transmission of RP: autosomal dominant (ADRP), autosomal recessive (arRP), X-linked (XLRP) and mitochondrial. The genetics behind the different forms of RP and the degree of severity vary, although some overlap, thus contributing to the difficulty of differential diagnosis. RP can manifest either as a non-syndromic disease, or as part of a syndrome, such as in Usher’s syndrome (hearing and vision loss) and BardetBiedl syndrome (a ciliopathy). The purpose of this review is to summarize the major genetic and molecular findings, as well as the diseases, associated with RP. Due to space limitations, this review is not fully comprehensive. Keywords: Retinitis pigmentosa, non-syndromic retinitis pigmentosa, rod-cone dystrophy, rhodopsin Introduction Retinitis pigmentosa (RP) is a heterogeneous set of inherited retinal diseases. -

Albinism Terminology
Albinism Terminology Oculocutaneous Albinism (OCA): Oculocutaneous (pronounced ock-you-low-kew- TAIN-ee-us) Albinism is an inherited genetic condition characterized by the lack of or diminished pigment in the hair, skin, and eyes. Implications of this condition include eye and skin sensitivities to light and visual impairment. Ocular Albinism (OA): Ocular Albinism is an inherited genetic condition, diagnosed predominantly in males, characterized by the lack of pigment in the eyes. Implications of this condition include eye sensitivities to light and visual impairment. Hermansky Pudlak Syndrome (HPS): Hermansky-Pudlak Syndrome is a type of albinism which includes a bleeding tendency and lung disease. HPS may also include inflammatory bowel disease or kidney disease. The severity of these problems varies much from person to person, and the condition can be difficult to diagnose with traditional blood tests Chediak Higashi Syndrome: Chediak Higashi Syndrome is a type of albinism in which the immune system is affected. Illnesses and infections are common from infancy and can be severe. Issues also arise with blood clotting and severe bleeding. Melanin: Melanin is pigment found in a group of cells called melanocytes in most organisms. In albinism, the production of melanin is impaired or completely lacking. Nystagmus: Nystagmus is an involuntary movement of the eyes in either a vertical, horizontal, pendular, or circular pattern caused by a problem with the visual pathway from the eye to the brain. As a result, both eyes are unable to hold steady on objects being viewed. Nystagmus may be accompanied by unusual head positions and head nodding in an attempt to compensate for the condition. -

Stargardt's Hereditary Progressive Macular Degeneration
Brit. 7. Ophthal. ( 1972) 56, 8I 7 Br J Ophthalmol: first published as 10.1136/bjo.56.11.817 on 1 November 1972. Downloaded from Stargardt's hereditary progressive macular degeneration A. RODMAN IRVINE AND FLOYD L. WERGELAND, JR From Ophthalmology Service, Letterman General Hospital, Presidio of San Francisco, California In a series of three papers, Stargardt ( 1909,I93, I9I6) described with precision and thoroughness a form of hereditary macular degeneration which has become known as Stargardt's disease. The purposes of the present paper are to review Stargardt's original observations, to argue that Stargardt's disease and fundus flavimaculatus with atrophic macular degeneration are identical, and to present a series of patients studied by fluorescein angiography consistent with the hypothesis that a late secondary or disuse atrophy of the choriocapillaris occurs in Stargardt's disease. Stargardt described four families. The disease seemed to show recessive inheritance, being present in siblings but never in successive generations. Symptoms were usually first noted between 8 and I6 years of age, and then progressed gradually and inexorably until all macular function was destroyed some years later. The first finding was a decrease in visual acuity, often more noticeable in the bright light than in the dark, with minimal by copyright. ophthalmoscopic changes. A faint irregularity in the pigment in the macular region was often all that could be seen at this stage, and the fundus might easily be passed as normal. Later, the foveal reflex was lost. Soft yellow-grey spots appeared in the macula which initially were barely distinguishable from the fundus background. -

Stargardt Disease
Stargardt disease Author: Professor August. F. Deutman1 Creation Date: January 2003 Scientific Editor: Professor Jean-Jacques De Laey 1Institute of Ophthalmology, University Hospital Nijmegen, Postbox 9101, 6500 HB Nijmegen, Netherlands. Abstract Keywords Disease name and synonyms Excluded diseases Definition Frequency Clinical description Management including treatment Etiology Diagnosis References Abstract Stargardt's disease is a form of juvenile hereditary macular degeneration characterized by discrete yellowish round or pisciform flecks around the macula at the level of the retinal pigment epithelium (rpe). Stargardt's disease is the most common hereditary macular dystrophy. Prevalence is estimated between 1 in 8,000 and 1 in 10,000. Disease onset occurs typically in the first or second decade of life and manifests as decreased visual acuity. In the early stages, the macula usually shows discrete rpe changes, followed later by an horizontal ovoid zone of beaten bronze atrophy. In final stages, the macula can be associated with central areolar choroidal dystrophy. Fluorescein angiography reveals the characteristic dark choroid (''silence choroidien''), which probably results from the accumulation of lipofuscin in the rpe. This disease has usually an autosomal recessive inheritance pattern but some dominant pedigrees have been reported. The autosomal type has been associated with mutations in the ABCR gene, which encodes a transmembrane transporter protein expressed by the rod outer segments. There is currently no treatment available for Stargardt's disease. Keywords Stargardt, Macula, Fundus flavimaculatus Disease name and synonyms Definition • Stargardt’s disease Stargardt’s disease (Stargardt, 1909, 1913, • Fundus flavimaculatus 1916, 1917, 1925; Weleber, 1994; Armstrong et al., 1998) is a form of juvenile hereditary macular Excluded diseases degeneration characterized by discrete yellowish • Cone dystrophy round or pisciform flecks around the macula at the level of the retinal pigment epithelium (rpe). -

Colour Vision Deficiency
Eye (2010) 24, 747–755 & 2010 Macmillan Publishers Limited All rights reserved 0950-222X/10 $32.00 www.nature.com/eye Colour vision MP Simunovic REVIEW deficiency Abstract effective "treatment" of colour vision deficiency: whilst it has been suggested that tinted lenses Colour vision deficiency is one of the could offer a means of enabling those with commonest disorders of vision and can be colour vision deficiency to make spectral divided into congenital and acquired forms. discriminations that would normally elude Congenital colour vision deficiency affects as them, clinical trials of such lenses have been many as 8% of males and 0.5% of femalesFthe largely disappointing. Recent developments in difference in prevalence reflects the fact that molecular genetics have enabled us to not only the commonest forms of congenital colour understand more completely the genetic basis of vision deficiency are inherited in an X-linked colour vision deficiency, they have opened the recessive manner. Until relatively recently, our possibility of gene therapy. The application of understanding of the pathophysiological basis gene therapy to animal models of colour vision of colour vision deficiency largely rested on deficiency has shown dramatic results; behavioural data; however, modern molecular furthermore, it has provided interesting insights genetic techniques have helped to elucidate its into the plasticity of the visual system with mechanisms. respect to extracting information about the The current management of congenital spectral composition of the visual scene. colour vision deficiency lies chiefly in appropriate counselling (including career counselling). Although visual aids may Materials and methods be of benefit to those with colour vision deficiency when performing certain tasks, the This article was prepared by performing a evidence suggests that they do not enable primary search of Pubmed for articles on wearers to obtain normal colour ‘colo(u)r vision deficiency’ and ‘colo(u)r discrimination. -

1 Human Color Vision
CAMC01 9/30/04 3:13 PM Page 1 1 Human Color Vision Color appearance models aim to extend basic colorimetry to the level of speci- fying the perceived color of stimuli in a wide variety of viewing conditions. To fully appreciate the formulation, implementation, and application of color appearance models, several fundamental topics in color science must first be understood. These are the topics of the first few chapters of this book. Since color appearance represents several of the dimensions of our visual experience, any system designed to predict correlates to these experiences must be based, to some degree, on the form and function of the human visual system. All of the color appearance models described in this book are derived with human visual function in mind. It becomes much simpler to understand the formulations of the various models if the basic anatomy, physiology, and performance of the visual system is understood. Thus, this book begins with a treatment of the human visual system. As necessitated by the limited scope available in a single chapter, this treatment of the visual system is an overview of the topics most important for an appreciation of color appearance modeling. The field of vision science is immense and fascinating. Readers are encouraged to explore the liter- ature and the many useful texts on human vision in order to gain further insight and details. Of particular note are the review paper on the mechan- isms of color vision by Lennie and D’Zmura (1988), the text on human color vision by Kaiser and Boynton (1996), the more general text on the founda- tions of vision by Wandell (1995), the comprehensive treatment by Palmer (1999), and edited collections on color vision by Backhaus et al.