Glucocorticoids Suppress Wnt16 Expression in Osteoblasts in Vitro
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WNT16-Expressing Acute Lymphoblastic Leukemia Cells Are Sensitive to Autophagy Inhibitors After ER Stress Induction
ANTICANCER RESEARCH 35: 4625-4632 (2015) WNT16-expressing Acute Lymphoblastic Leukemia Cells are Sensitive to Autophagy Inhibitors after ER Stress Induction MELETIOS VERRAS1, IOANNA PAPANDREOU2 and NICHOLAS C. DENKO2 1General Biology Laboratory, School of Medicine, University of Patra, Rio, Greece; 2Department of Radiation Oncology, Wexner Medical Center and Comprehensive Cancer Center, The Ohio State University, Columbus OH, U.S.A. Abstract. Background: Previous work from our group showed burden of proteins in the ER through decreased translation, hypoxia can induce endoplasmic reticulum (ER) stress and increased chaperone expression, and increased removal of the block the processing of the WNT3 protein in cells engineered malfolded proteins through degradation. If the cell is unable to express WNT3a. Acute lymphoblastic leukemia (ALL) cells to relieve the ER stress, then cellular death can ensue (3). with the t(1:19) translocation express the WNT16 gene, which The microenvironment of solid tumors is often poorly is thought to contribute to transformation. Results: ER-stress perfused, resulting in regions of hypoxia and nutrient blocks processing of endogenous WNT16 protein in RCH-ACV deprivation (4, 5). However, hypoxia has been also shown to and 697 ALL cells. Biochemical analysis showed an impact cancer of the bone marrow such as aggressive aggregation of WNT16 proteins in the ER of stressed cells. leukemia (6). In addition to inducing the hypoxia-inducible These large protein masses cannot be completely cleared by factor 1 (HIF1) transcription factor, severe hypoxia induces ER-associated protein degradation, and require for additional stress in the ER (7, 8). Cells with compromised ability to autophagic responses. -

Altered Expression of PRKX, WNT3 and WNT16 in Human Nodular
ANTICANCER RESEARCH 36 : 4545-4552 (2016) doi:10.21873/anticanres.11002 Altered Expression of PRKX , WNT3 and WNT16 in Human Nodular Basal Cell Carcinoma NATALIA GURGEL DO CARMO 1,2 , LUIS HENRIQUE TOSHIHIRO SAKAMOTO 3, ROBERT POGUE 1, CINTIA DO COUTO MASCARENHAS 1, SIMONE KARST PASSOS 4, MARIA SUELI SOARES FELIPE 1,2 and ROSÂNGELA VIEIRA DE ANDRADE 1 1Program of Genomic Sciences and Biotechnology, Catholic University of Brasília, Brasília, DF, Brazil; 2Program of Molecular Pathology, University of Brasília, Brasília, DF, Brazil; 3Domingos A. Boldrini Center for Hematological Investigation, Campinas, SP, Brazil; 4Dermatology Service, Asa Norte Regional Hospital, Brasília, DF, Brazil Abstract. Background/Aim: Nodular and superficial are the differences in their biological behavior, such as tumor most common subtypes of basal cell carcinoma (BCC). growth pattern, potential for recurrence and metastasis, Signaling pathways such as Hedgehog (HH) and Wingless histological pattern and genetic factors. In addition, it is (WNT) signaling are associated with BCC phenotypic important to consider extrinsic factors, such as site of origin, variation. The aim of the study was to evaluate of the therapeutic choice and immunological state of the person expression profiles of 84 genes related to the WNT and HH with the tumor (3). Nodular BCC is the most common signaling pathways in patients with nodular and superficial biopsied subtype; it usually manifests as a single lesion and BCC. Materials and Methods: A total of 58 BCCs and 13 mostly affects head and neck areas (8). Histologically, the samples of normal skin were evaluated by quantitative real- tumor is a well-defined structure with precise contours; it time polymerase chain reaction (qPCR) to detect the gene- presents basaloid cells of nodular mass separated from the expression profile. -

The Draft Genomes of Softshell Turtle and Green Sea Turtle Yield Insights
LETTERS OPEN The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan Zhuo Wang1,12, Juan Pascual-Anaya2,12, Amonida Zadissa3,12, Wenqi Li4,12, Yoshihito Niimura5, Zhiyong Huang1, Chunyi Li4, Simon White3, Zhiqiang Xiong1, Dongming Fang1, Bo Wang1, Yao Ming1, Yan Chen1, Yuan Zheng1, Shigehiro Kuraku2, Miguel Pignatelli6, Javier Herrero6, Kathryn Beal6, Masafumi Nozawa7, Qiye Li1, Juan Wang1, Hongyan Zhang4, Lili Yu1, Shuji Shigenobu7, Junyi Wang1, Jiannan Liu4, Paul Flicek6, Steve Searle3, Jun Wang1,8,9, Shigeru Kuratani2, Ye Yin4, Bronwen Aken3, Guojie Zhang1,10,11 & Naoki Irie2 The unique anatomical features of turtles have raised Three major hypotheses have been proposed for the evolutionary unanswered questions about the origin of their unique body origin of turtles, including that they (i) constitute early-diverged rep- plan. We generated and analyzed draft genomes of the soft- tiles, called anapsids3, (ii) are a sister group of the lizard-snake-tuatara shell turtle (Pelodiscus sinensis) and the green sea turtle (Lepidosauria) clade4 or (iii) are closely related to a lineage that (Chelonia mydas); our results indicated the close relationship includes crocodilians and birds (Archosauria)5–8. Even using molecular of the turtles to the bird-crocodilian lineage, from which they approaches, inconsistency still remains6–9. To clarify the evolution of split ~267.9–248.3 million years ago (Upper Permian to Triassic). the turtle-specific body plan, we first addressed the question of evolu- We also found extensive expansion of olfactory receptor genes tionary origin of the turtle by performing the first genome-wide phylo- in these turtles. -

Epigenetic Modifiers DNMT3A and BCOR Are Recurrently Mutated In
ARTICLE https://doi.org/10.1038/s41467-019-12746-w OPEN Epigenetic modifiers DNMT3A and BCOR are recurrently mutated in CYLD cutaneous syndrome Helen R. Davies1,2,3, Kirsty Hodgson4, Edward Schwalbe5,6, Jonathan Coxhead4, Naomi Sinclair4, Xueqing Zou1,2,3, Simon Cockell4, Akhtar Husain7, Serena Nik-Zainal 1,2,3* & Neil Rajan4,8* Patients with CYLD cutaneous syndrome (CCS; syn. Brooke-Spiegler syndrome) carry germline mutations in the tumor suppressor CYLD and develop multiple skin tumors with 1234567890():,; diverse histophenotypes. Here, we comprehensively profile the genomic landscape of 42 benign and malignant tumors across 13 individuals from four multigenerational families and discover recurrent mutations in epigenetic modifiers DNMT3A and BCOR in 29% of benign tumors. Multi-level and microdissected sampling strikingly reveal that many clones with different DNMT3A mutations exist in these benign tumors, suggesting that intra-tumor heterogeneity is common. Integrated genomic, methylation and transcriptomic profiling in selected tumors suggest that isoform-specific DNMT3A2 mutations are associated with dysregulated methylation. Phylogenetic and mutational signature analyses confirm cylin- droma pulmonary metastases from primary skin tumors. These findings contribute to existing paradigms of cutaneous tumorigenesis and metastasis. 1 Wellcome Trust Sanger Institute, Hinxton, UK. 2 Academic Department of Medical Genetics, University of Cambridge, Cambridge, UK. 3 MRC Cancer Unit, University of Cambridge, Cambridge, UK. 4 Institute of Genetic Medicine, Newcastle University, Newcastle upon Tyne, UK. 5 Department of Applied Sciences, Northumbria University, Newcastle upon Tyne, UK. 6 Northern Institute for Cancer Research, Newcastle University, Newcastle upon Tyne, UK. 7 Department of Pathology, Royal Victoria Infirmary, Newcastle upon Tyne, UK. 8 Department of Dermatology, Royal Victoria Infirmary, Newcastle upon Tyne, UK. -

WNT Signaling As a Therapeutic Target for Glioblastoma
International Journal of Molecular Sciences Review WNT Signaling as a Therapeutic Target for Glioblastoma Michael Latour 1,†, Nam-Gu Her 2,†, Santosh Kesari 1 and Elmar Nurmemmedov 1,* 1 Saint John’s Cancer Institute at Providence Saint John’s Health Center, Santa Monica, CA 90404, USA; [email protected] (M.L.); [email protected] (S.K.) 2 Samsung Medical Center, Seoul 135-710, Korea; [email protected] * Correspondence: [email protected] † Equally contributing author. Abstract: The WNT (Wingless/Integrated) signaling pathway is implicated in various stages of glioblastoma, which is an aggressive brain tumor for which therapeutic options are limited. WNT has been recognized as a hallmark of therapeutic challenge due to its context-dependent role and critical function in healthy tissue homeostasis. In this review, we deeply scrutinize the WNT signaling pathway and its involvement in the genesis of glioblastoma as well as its acquired therapy resistance. We also provide an analysis of the WNT pathway in terms of its therapeutic importance in addition to an overview of the current targeted therapies under clinical investigation. Keywords: glioblastoma; WNT; beta-catenin; therapeutic; drug; resistance 1. Biology of WNT Signaling WNT (Wingless/Integrated) signaling regulates key cellular events during the devel- opment of the central nervous system. Particularly, it regulates self-renewal, differentiation, Citation: Latour, M.; Her, N.-G.; migration and signaling of neural stem cells in the fetal ventricular zone, the postnatal Kesari, S.; Nurmemmedov, E. WNT subventricular zone and the hippocampus [1,2]. It has been abundantly demonstrated that Signaling as a Therapeutic Target for hyperactivation of WNT signaling is associated with driving malignant transformation and Glioblastoma. -

Integrative Genomic Analyses of WNT11: Transcriptional Mechanisms Based on Canonical WNT Signals and GATA Transcription Factors
247-251 17/6/2009 01:23 ÌÌ Page 247 INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 24: 247-251, 2009 247 Integrative genomic analyses of WNT11: Transcriptional mechanisms based on canonical WNT signals and GATA transcription factors MASUKO KATOH1 and MASARU KATOH2 1M&M Medical BioInformatics, Hongo 113-0033; 2Genetics and Cell Biology Section, National Cancer Center, Tokyo 104-0045, Japan Received April 22, 2009; Accepted May 29, 2009 DOI: 10.3892/ijmm_00000227 Abstract. We and others previously cloned and characterized activates the Ca2+-MAP3K7-NLK signaling cascade to break vertebrate WNT11 orthologs, which are involved in gastrulation, the canonical WNT signaling. Canonical WNT-to-WNT11 neurulation, cardiogenesis, nephrogenesis, and chondrogenesis signaling loop is involved in cellular migration during embryo- during fetal development. WNT11 orthologs activate both genesis as well as tumor invasion during carcinogenesis. canonical and non-canonical WNT signaling cascades depending on the expression profile of WNT receptors, such Introduction as Frizzled family members, LRP6, ROR2, and RYK. Human WNT11 is expressed in breast cancer, gastric cancer, esophageal WNT family members are secreted proteins with glycolipid cancer, colorectal cancer, neuroblastoma, Ewing sarcoma, and modifications, which are involved in embryogenesis, adult- prostate cancer. Canonical WNT signals and GATA family tissue homeostasis, and carcinogenesis (1-5). WNT1, WNT2, members are involved in WNT11 transcription during embryo- WNT2B, WNT3, WNT3A, WNT4, WNT5A, WNT5B, WNT6, genesis of model animals; however, precise mechanisms of WNT7A, WNT7B, WNT8A, WNT8B, WNT9A, WNT9B, WNT11 expression remain unclear. Here, refined integrative WNT10A, WNT10B, WNT11, and WNT16 genes are conserved genomic analyses of WNT11 are carried out to elucidate the in the mammalian genomes (6), whereas additional wnt family mechanisms of WNT11 transcription. -

FGF Signalling Specifies Haematopoietic Stem Cells Through Its Regulation of Somitic Notch Signalling
ARTICLE Received 26 Mar 2014 | Accepted 16 Oct 2014 | Published 27 Nov 2014 DOI: 10.1038/ncomms6583 FGF signalling specifies haematopoietic stem cells through its regulation of somitic Notch signalling Yoonsung Lee1, Jennifer E. Manegold1, Albert D. Kim1, Claire Pouget1, David L. Stachura1,2, Wilson K. Clements1,3 & David Traver1 Haematopoietic stem cells (HSCs) derive from haemogenic endothelial cells of the primitive dorsal aorta (DA) during vertebrate embryogenesis. The molecular mechanisms governing this unique endothelial to haematopoietic transition remain unclear. Here, we demonstrate a novel requirement for fibroblast growth factor (FGF) signalling in HSC emergence. This requirement is non-cell-autonomous, and acts within the somite to bridge the Wnt and Notch signalling pathways. We previously demonstrated that Wnt16 regulates the somitic expres- sion of two Notch ligands, deltaC (dlc) and deltaD (dld), whose combined function is required for HSC fate. How Wnt16 connects to Notch function has remained an open question. Our current studies demonstrate that FGF signalling, via FGF receptor 4 (Fgfr4), mediates a signal- transduction pathway between Wnt16 and Dlc, but not Dld, to regulate HSC specification. Our findings demonstrate that FGF signalling acts as a key molecular relay within the developmental HSC niche to instruct HSC fate. 1 Department of Cellular and Molecular Medicine and Section of Cell and Developmental Biology, University of California, San Diego, La Jolla, California 92093, USA. 2 Department of Biological Sciences, California State University, Chico, California 95929, USA. 3 Department of Hematology, St Jude Children’s Research Hospital, Memphis, Tennessee 38105, USA. Correspondence and requests for materials should be addressed to D.T. -

Induction of WNT16 Via Peptide-Mrna Nanoparticle-Based Delivery Maintains Cartilage Homeostasis
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2020 Induction of WNT16 via peptide-mRNA nanoparticle-based delivery maintains cartilage homeostasis Huimin Yan Ying Hu Antonina Akk Muhammad Farooq Rai Hua Pan See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Authors Huimin Yan, Ying Hu, Antonina Akk, Muhammad Farooq Rai, Hua Pan, Samuel A Wickline, and Christine T N Pham pharmaceutics Article Induction of WNT16 via Peptide-mRNA Nanoparticle-Based Delivery Maintains Cartilage Homeostasis Huimin Yan 1,2, Ying Hu 1,2, Antonina Akk 1, Muhammad Farooq Rai 3 , Hua Pan 4, Samuel A. Wickline 4,* and Christine T.N. Pham 1,2,* 1 Department of Medicine, Washington University School of Medicine, St. Louis, MO 63110, USA; [email protected] (H.Y.); [email protected] (Y.H.); [email protected] (A.A.) 2 John Cochran Veterans Affairs Medical Center, St. Louis, MO 63106, USA 3 Department of Orthopaedic Surgery, Washington University School of Medicine, St. Louis, MO 63110, USA; [email protected] 4 Department of Cardiovascular Sciences, University of South Florida, Tampa, FL 33620, USA; [email protected] * Correspondence: [email protected] (S.A.W.); [email protected] (C.T.N.P.) Received: 21 November 2019; Accepted: 13 January 2020; Published: 17 January 2020 Abstract: Osteoarthritis (OA) is a progressive joint disease that causes significant disability and pain and for which there are limited treatment options. We posit that delivery of anabolic factors that protect and maintain cartilage homeostasis will halt or retard OA progression. We employ a peptide-based nanoplatform to deliver Wingless and the name Int-1 (WNT) 16 messenger RNA (mRNA) to human cartilage explants. -
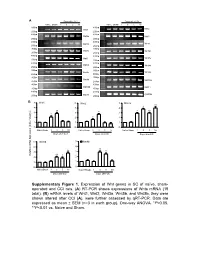
Supplementary Figure 1. Expression of Wnt Genes in SC of Naïve
A Days after CCI Days after CCI Naive Sham 1 3 5 10 Naive Sham 1 3 5 10 500bp 400bp Wnt1 Wnt2 250bp 250bp 400bp 400bp Wnt2b Wnt3 250bp 250bp 400bp 400bp Wnt3a Wnt4 250bp 250bp 300bp 400bp Wnt5a Wnt5b 200bp 250bp 400bp 400bp Wnt6 Wnt7a 250bp 250bp 400bp 400bp Wnt7b Wnt8a 250bp 250bp 400bp 350bp Wnt8b Wnt9a 250bp 200bp 400bp 400bp Wnt9b Wnt10a 250bp 250bp 400bp 400bp Wnt10b Wnt11 250bp 250bp 400bp 400bp Wnt16 GAPDH 250bp 250bp B 5 Wnt1 5 Wnt2 5 Wnt3a ** 4 4 4 * ** ** 3 3 * 3 * * 2 2 * 2 1 1 1 0 0 0 NaïveSham 1 3 5 10 Naïve Sham 1 3 5 10 Naïve Sham 1 3 5 10 Days after CCI Days after CCI Days after CCI 5 Wnt5b 5 Wnt8b 4 4 ** 3 ** 3 ** Relative mRNA Expression (fold of change) ** 2 * 2 1 1 0 0 NaïveSham 1 3 5 10 NaïveSham 1 3 5 10 Days after CCI Days after CCI Supplementary Figure 1 A B Negative Weak Moderate Strong Negative Weak Moderate Strong Large-sized cells 100% 100% 80% 80% 60% 60% 40% 20% 40% 0% 20% Sham CCI-1d CCI-14d Medium-sized cells Wnt3a immunoreactivity cells 0% 100% CGRP(+) IB4(+) 80% Small cells 60% 40% 20% 0% Sham CCI-1d CCI-14d Wnt3a immunoreactivity cells Small cells 100% 80% 60% 40% 20% 0% Sham CCI-1d CCI-14d Fig. S2 4 Fz1 4 Fz3 4 Fz4 4 Fz5 ** 3 3 ** 3 3 2 ** 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1 5 10 NaïveSham 1 5 10 Days after CCI Days after CCI Days after CCI Days after CCI 4 Fz6 4 Fz7 4 Fz8 4 Fz9 (Fold of Change) of (Fold 3 3 3 ** 3 Relative mRNA Expression Expression mRNA Relative ** 2 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1d 5d 10d NaïveSham 1 -

WNT Inhibitory Activity of Malus Pumila Miller Cv
nutrients Article WNT Inhibitory Activity of Malus Pumila miller cv Annurca and Malus domestica cv Limoncella Apple Extracts on Human Colon-Rectal Cells Carrying Familial Adenomatous Polyposis Mutations Gennaro Riccio 1 ID , Maria Maisto 1, Sara Bottone 1, Nadia Badolati 1, Giovanni Battista Rossi 2, Gian Carlo Tenore 1 ID , Mariano Stornaiuolo 1,* ID and Ettore Novellino 1,* 1 Department of Pharmacy, University of Naples Federico II, 80131 Naples, Italy; [email protected] (G.R.); [email protected] (M.M.); [email protected] (S.B.); [email protected] (N.B.); [email protected] (G.C.T.) 2 Gastroenterology and Gastrointestinal Endoscopy Unit, Istituto Nazionale Tumori-IRCCS-Fondazione G. Pascale, 80131 Naples, Italy; [email protected] * Correspondence: [email protected] (M.S.); [email protected] (E.N.); Tel.: +39-081-670-117 (M.S.); +39-081-678-643 (E.N.) Received: 2 October 2017; Accepted: 15 November 2017; Published: 18 November 2017 Abstract: Inhibitors of the Wingless-related Integration site (WNT)/β-catenin pathway have recently been under consideration as potential chemopreventive agents against Familial Adenomatous Polyposis (FAP). This autosomal-dominant syndrome is caused by germline mutations in the gene coding for the protein APC and leads to hyperactivation of the WNT/β-catenin signaling pathway, uncontrolled intestinal cell proliferation and formation of adenocarcinomas. The aim of the present work was to: (i) test, on in vitro cultures of cells carrying FAP mutations and on ex vivo biopsies of FAP patients, the WNT inhibitory activity of extracts from two common southern Italian apples, Malus pumila Miller cv. -

The Impact of Rare and Low-Frequency Genetic Variants in Common Disease Lorenzo Bomba1, Klaudia Walter1 and Nicole Soranzo1,2,3*
Bomba et al. Genome Biology (2017) 18:77 DOI 10.1186/s13059-017-1212-4 REVIEW Open Access The impact of rare and low-frequency genetic variants in common disease Lorenzo Bomba1, Klaudia Walter1 and Nicole Soranzo1,2,3* of study participants [9–12]. Despite the increases in statis- Abstract tical power afforded by these large-scale studies, for the ma- Despite thousands of genetic loci identified to date, a jority of human traits genetic associations discovered large proportion of genetic variation predisposing to account for a fraction of disease or trait heritability (the complex disease and traits remains unaccounted for. “missing heritability” paradigm). Genetic variants that are Advances in sequencing technology enable focused outside the reach of the most statistically powered associ- explorations on the contribution of low-frequency and ation studies [13] are thought to contribute to the missing rare variants to human traits. Here we review heritability of many human traits, including common vari- experimental approaches and current knowledge on ants (here denoted by minor allele frequency [MAF] >5%) the contribution of these genetic variants in complex of very weak effect, low-frequency (MAF 1–5%) and rare disease and discuss challenges and opportunities for variants (MAF <1%) of small to modest effect, or a combin- personalised medicine. ation of both, with several possible scenarios all deemed plausibleinsimulationstudies[14]. Empirical studies attempting to understand the impact Introduction of rare or less common variation on human complex dis- Genetic research has played an instrumental role in the eases and traits remain to date relatively limited [15, 16], discovery of new biological pathways underpinning com- but some lessons on their properties are beginning to plex human disease and the evaluation of new targets for emerge from exome-wide and genome-wide sequencing therapeutic development. -

WNT16 Antagonises Excessive Canonical WNT Activation and Protects Cartilage in Osteoarthritis
Basic and translational research Ann Rheum Dis: first published as 10.1136/annrheumdis-2015-208577 on 4 May 2016. Downloaded from EXTENDED REPORT WNT16 antagonises excessive canonical WNT activation and protects cartilage in osteoarthritis Giovanna Nalesso,1 Bethan Lynne Thomas,1 Joanna Claire Sherwood,1,2 Jing Yu,3 Olga Addimanda,4,5 Suzanne Elizabeth Eldridge,1 Anne-Sophie Thorup,1 Leslie Dale,6 Georg Schett,7 Jochen Zwerina,7 Noha Eltawil,1,8 Costantino Pitzalis,1 Francesco Dell’Accio1 Handling editor Tore K Kvien ABSTRACT the β-catenin-dependent (or canonical WNT) ▸ Additional material is Objective Both excessive and insufficient activation of pathway, engagement of WNTs with their receptors published online only. To view WNT signalling results in cartilage breakdown and results in the accumulation and nuclear translocation please visit the journal online osteoarthritis. WNT16 is upregulated in the articular of β-catenin and transcriptional activation of target (http://dx.doi.org/10.1136/ 12 annrheumdis-2015-208577). cartilage following injury and in osteoarthritis. Here, we genes. The non-canonical pathways are less under- investigate the function of WNT16 in osteoarthritis and stood and involve Ca2+ signalling, phosphatidylinosi- the downstream molecular mechanisms. tol-4,5-biphosphate 3-kinase (PI3K) and activation of For numbered affiliations see Methods Osteoarthritis was induced by destabilisation of cjun N-terminal kinase (JNK).13 end of article. the medial meniscus in wild-type and WNT16-deficient Although genetic studies in humans and experi- Correspondence to mice. Molecular mechanisms and downstream effects were mental studies in animals provide overwhelming evi- Dr Bethan Lynne Thomas, studiedinvitroandinvivoinprimarycartilageprogenitor dence for the role of WNT signalling in cartilage Barts and the London School cells and primary chondrocytes.