Instructional Template for Therapeutic Protocol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Pilot Trial of Clofarabine Added to Standard Busulfan and Fludarabine for Conditioning Prior to Allogeneic Hematopoietic Cell Transplantation
NCT# 01596699 A Pilot Trial of Clofarabine Added to Standard Busulfan and Fludarabine for Conditioning Prior to Allogeneic Hematopoietic Cell Transplantation Protocol Number: CC #110819 Version Number: 7.0 Version Date: June 6, 2013 Study Drug: Clofarabine Principal Investigator (Sponsor-Investigator) Christopher C. Dvorak, MD Co-Investigators Janel Long-Boyle, PharmD, PhD Morton Cowan, MD Biljana Horn, MD James Huang, MD Robert Goldsby, MD Kristin Ammon, MD Justin Wahlstrom, MD Statistician Stephen Shiboski, PhD Clinical Research Coordinator Revision History Version #1.0 9/19/11 Version #2.0 11/8/11 Version #3.1 02/23/12 Version #4.0 04/10/12 Version #5.0 04/30/12 Version #6.0 08/09/12 Version #7.0 06/07/13 Clofarabine Version 4.0, 04/10/12 Page 1 of 33 1.0 Hypothesis and Specific Aims: 1.1 Hypothesis: The addition of Clofarabine to standard Busulfan and Fludarabine prior to allogeneic hematopoietic cell transplant (HCT) will be safe and will improve engraftment rates of children with non-malignant diseases or decrease relapse rates of patients with high-risk myeloid malignancies. 1.2 Specific Aims: 1.2.1 Primary Objective: To determine the toxicity of the addition of Clofarabine to a conditioning backbone of Busulfan plus Fludarabine in patients with myeloid malignancies and non-malignant diseases undergoing allogeneic HCT. 1.2.2 Secondary Objectives: a. To determine if the addition of Clofarabine to a conditioning backbone of Busulfan plus Fludarabine improves the engraftment rate of patients with non-malignant diseases (Stratum A) undergoing allogeneic HCT as compared to historic controls. -

214120Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 214120Orig1s000 MULTI-DISCIPLINE REVIEW Summary Review Office Director Cross Discipline Team Leader Review Clinical Review Non-Clinical Review Statistical Review Clinical Pharmacology Review NDA Multidisciplinary Review and Evaluation Application Number NDA 214120 Application Type Type 3 Priority or Standard Priority Submit Date 3/3/2020 Received Date 3/3/2020 PDUFA Goal Date 9/3/2020 Office/Division OOD/DHM1 Review Completion Date 9/1/2020 Applicant Celgene Corporation Established Name Azacitidine (Proposed) Trade Name Onureg Pharmacologic Class Nucleoside metabolic inhibitor Formulations Tablet (200 mg, 300 mg) (b) (4) Applicant Proposed Indication/Population Recommendation on Regulatory Regular approval Action Recommended Indication/ For continued treatment of adult patients with acute Population myeloid leukemia who achieved first complete remission (CR) or complete remission with incomplete blood count recovery (CRi) following intensive induction chemotherapy and are not able to complete intensive curative therapy. SNOMED CT for the Recommended 91861009 Indication/Population Recommended Dosing Regimen 300 mg orally daily on Days 1 through 14 of each 28-day cycle Reference ID: 4664570 NDA Multidisciplinary Review and Evaluation NDA 214120 Onureg (azacitidine tablets) TABLE OF CONTENTS TABLE OF CONTENTS ................................................................................................................................... 2 TABLE OF TABLES ....................................................................................................................................... -
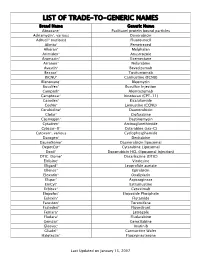
Trade-To-Generic Names
LIST OF TRADE-TO-GENERIC NAMES Brand Name Generic Name Abraxane® Paclitaxel protein bound particles Adriamycin®, various Doxorubicin Adrucil® (various) Fluorouracil Alimta® Pemetrexed Alkeran® Melphalan Arimidex® Anastrozole Aromasin® Exemestane Arranon® Nelarabine Avastin® Bevacizumab Bexxar® Tositumomab BiCNU® Carmustine (BCNU) Blenoxane® Bleomycin Busulfex® Busulfan Injection Campath® Alemtuzumab Camptosar® Irinotecan (CPT-11) Casodex® Bicalutamide CeeNu® Lomustine (CCNU) Cerubidine® Daunorubicin Clolar® Clofarabine Cosmegen® Dactinomycin Cytadren® Aminoglutethimide Cytosar-U® Cytarabine (ara-C) Cytoxan®, various Cyclophosphamide Dacogen® Decitabine DaunoXome® Daunorubicin liposomal DepotCyt® Cytarabine Liposomal Doxil® Doxorubicin HCL (liposomal injection) DTIC-Dome® Dacarbazine (DTIC) Eldisine® Vindesine Eligard® Leuprolide acetate Ellence® Epirubicin Eloxatin® Oxaliplatin Elspar® Asparaginase EmCyt® Estramustine Erbitux® Cetuximab Etopofos® Etoposide Phosphate Eulexin® Flutamide Fareston® Toremifene Faslodex® Fluvestrant Femara® Letrozole Fludara® Fludarabine Gemzar® Gemcitabine Gleevec® Imatinib Gliadel® Carmustine Wafer Halotestin® Fluoxymesterone Last Updated on January 15, 2007 Brand Name Generic Name Herceptin® Trastuzumab Hexalen® Altretamine Hycamtin® Topotecan Hydrea® Hydroxyurea Idamycin® Idarubicin Ifex® Ifosfamide Intron A® Interferon alfa-2b Iressa® Gefitinib Leukeran® Chlorambucil Leukine® Sargramostim Leustatin® Cladribine Lupron depot® Leuprolide acetate depot Lupron® Leuprolide acetate Matulane® Procarbazine Megace® -

Clofarabine/Busulfan-Based Reduced Intensity
www.oncotarget.com Oncotarget, 2018, Vol. 9, (No. 93), pp: 36603-36612 Research Paper Clofarabine/busulfan-based reduced intensity conditioning regimens provides very good survivals in acute myeloid leukemia patients in complete remission at transplant: a retrospective study on behalf of the SFGM-TC Amandine Le Bourgeois1, Myriam Labopin2, Mathieu Leclerc3, Régis Peffault de Latour4, Jean-Henri Bourhis5, Patrice Ceballos6, Corentin Orvain7, Hélène Labussière Wallet8, Karin Bilger9, Didier Blaise10, Marie-Thérese Rubio11, Thierry Guillaume1, Mohamad Mohty2, Patrice Chevallier1 and on behalf of Société Francophone de Greffe de Moelle et de Thérapie Cellulaire 1Department of Hematology, CHU Hôtel Dieu, Nantes, France 2Department of Hematology, Hôpital Saint Antoine, Paris, France 3Department of Hematology, Hôpital Henri Mondor, Créteil, France 4Department of Hematology, Hôpital Saint Louis, Université Paris 7, Denis Diderot, Paris, France 5Department of Hematology, Hôpital Gustave Roussy, Paris, France 6Department of Hematology, CHU de Montpellier, Montpellier, France 7Department of Hematology, CHU d’Angers, Angers, France 8Department of Hematology, Centre Hospitalier Lyon Sud, Lyon, France 9Department of Hematology, CHU Strasbourg, Strasbourg, France 10Department of Hematology, Centre de Recherche en Cancérologie de Marseille, Institut Paoli Calmettes, Marseille, France 11Department of Hematology, CHU Nancy, Nancy, France Correspondence to: Amandine Le Bourgeois, email: [email protected] Patrice Chevallier, email: [email protected] Keywords: allogeneic stem cell transplantation; clofarabine; busulfan; reduced intensity conditioning regimen; acute myeloid leukemia Received: August 24, 2018 Accepted: November 01, 2018 Published: November 27, 2018 Copyright: Bourgeois et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. -

Clofarabine (Clolar®) (“Kloe-FAR-A-Been”)
Clofarabine (Clolar®) (“kloe-FAR-a-been”) How drug is given: by vein (IV) Purpose: To treat leukemia. Things that may occur during or within hours of each treatment • Changes in your pulse and blood pressure may occur. • You may have nausea, vomiting, and/or loss of appetite. Nausea and vomiting may begin soon after the drug is given and may last more than 24 hours. You may be given medicine to help with this. • It is important to drink extra fluids after receiving this medication. Things that may occur a few days to weeks later 1. If you start to feel joint pain, swelling, weakness or stiffness, call your cancer care team. 2. Loose stools or diarrhea may occur within a few days after the drug is started. You may take loperamide (Imodium A-D®) to help control diarrhea. You can buy this at most drug stores. Be sure to also drink more fluids (water, juice, sports drinks). If these do not help within 24 hours, call your cancer care team. 3. Some patients may feel very tired, also known as fatigue. You may need to rest or take naps more often. Mild to moderate exercise can also be helpful in maintaining your energy. 4. Your blood cell counts may drop. This is known as bone marrow suppression. This includes a decrease in your: • Red blood cells, which carry oxygen in your body to help give you energy • White blood cells, which fight infection in your body • Platelets, which help clot the blood to stop bleeding This may happen 7 to 14 days after the drug is given and then blood counts should return to normal. -

Thiopurine Drug Therapy
Thiopurine Drug Therapy Thiopurine drug therapy is used for autoimmune diseases, inflammatory bowel disease, acute lymphoblastic leukemia, and to prevent rejection after solid organ transplant. The inactivation of thiopurine drugs is catalyzed in part by thiopurine Tests to Consider methyltrasferase (TPMT) and nudix hydrolase 15 (NUDT15). Variants in the TPMT and/or NUDT15 genes are associated with an accumulation of cytotoxic metabolites Thiopurine Methyltransferase, RBC leading to increased risk of drug-related toxicity with standard doses of thiopurine 0092066 drugs, and the effects on thiopurine catabolism can be additive. Method: Enzymatic/Quantitative Liquid Chromatography-Tandem Mass Spectrometry The enzyme activity phenotype of TPMT can also be measured directly when Phenotype test to assess risk for severe performed prior to drug administration. Complementary to pretherapeutic tests, myelosuppression with standard dosing of concentrations of thiopurines and metabolites can be measured after initiation of thiopurine drugs therapy to optimize dose. Use for individuals being considered for thiopurine therapy Must be performed before thiopurine therapy is initiated Disease Overview Can also detect rapid metabolizer phenotype Prevalence TPMT and NUDT15 3001535 Method: Polymerase Chain Very low/absent TPMT activity: ~3/1,000 individuals Reaction/Fluorescence Monitoring Intermediate TPMT activity: ~10% of Caucasian individuals Normal TPMT activity: ~90% of individuals Genotyping test to assess genetic risk for severe myelosuppression -

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

The New Therapeutic Strategies in Pediatric T-Cell Acute Lymphoblastic Leukemia
International Journal of Molecular Sciences Review The New Therapeutic Strategies in Pediatric T-Cell Acute Lymphoblastic Leukemia Marta Weronika Lato 1 , Anna Przysucha 1, Sylwia Grosman 1, Joanna Zawitkowska 2 and Monika Lejman 3,* 1 Student Scientific Society, Laboratory of Genetic Diagnostics, Medical University of Lublin, 20-093 Lublin, Poland; [email protected] (M.W.L.); [email protected] (A.P.); [email protected] (S.G.) 2 Department of Pediatric Hematology, Oncology and Transplantology, Medical University of Lublin, 20-093 Lublin, Poland; [email protected] 3 Laboratory of Genetic Diagnostics, Medical University of Lublin, 20-093 Lublin, Poland * Correspondence: [email protected] Abstract: Childhood acute lymphoblastic leukemia is a genetically heterogeneous cancer that ac- counts for 10–15% of T-cell acute lymphoblastic leukemia (T-ALL) cases. The T-ALL event-free survival rate (EFS) is 85%. The evaluation of structural and numerical chromosomal changes is important for a comprehensive biological characterization of T-ALL, but there are currently no ge- netic prognostic markers. Despite chemotherapy regimens, steroids, and allogeneic transplantation, relapse is the main problem in children with T-ALL. Due to the development of high-throughput molecular methods, the ability to define subgroups of T-ALL has significantly improved in the last few years. The profiling of the gene expression of T-ALL has led to the identification of T-ALL subgroups, and it is important in determining prognostic factors and choosing an appropriate treatment. Novel therapies targeting molecular aberrations offer promise in achieving better first remission with the Citation: Lato, M.W.; Przysucha, A.; hope of preventing relapse. -

BC Cancer Protocol Summary for Treatment of Lymphoma with Dose- Adjusted Etoposide, Doxorubicin, Vincristine, Cyclophosphamide
BC Cancer Protocol Summary for Treatment of Lymphoma with Dose- Adjusted Etoposide, DOXOrubicin, vinCRIStine, Cyclophosphamide, predniSONE and riTUXimab with Intrathecal Methotrexate Protocol Code LYEPOCHR Tumour Group Lymphoma Contact Physician Dr. Laurie Sehn Dr. Kerry Savage ELIGIBILITY: One of the following lymphomas: . Patients with an aggressive B-cell lymphoma and the presence of a dual translocation of MYC and BCL2 (i.e., double-hit lymphoma). Histologies may include DLBCL, transformed lymphoma, unclassifiable lymphoma, and intermediate grade lymphoma, not otherwise specified (NOS). Patients with Burkitt lymphoma, who are not candidates for CODOXM/IVACR (such as those over the age of 65 years, or with significant co-morbidities) . Primary mediastinal B-cell lymphoma Ensure patient has central line EXCLUSIONS: . Cardiac dysfunction that would preclude the use of an anthracycline. TESTS: . Baseline (required before first treatment): CBC and diff, platelets, BUN, creatinine, bilirubin. ALT, LDH, uric acid . Baseline (required, but results do not have to be available to proceed with first treatment): results must be checked before proceeding with cycle 2): HBsAg, HBcoreAb, . Baseline (optional, results do not have to be available to proceed with first treatment): HCAb, HIV . Day 1 of each cycle: CBC and diff, platelets, (and serum bilirubin if elevated at baseline; serum bilirubin does not need to be requested before each treatment, after it has returned to normal), urinalysis for microscopic hematuria (optional) . Days 2 and 5 of each cycle (or days of intrathecal treatment): CBC and diff, platelets, PTT, INR . For patients on cyclophosphamide doses greater than 2000 mg: Daily urine dipstick for blood starting on day cyclophosphamide is given. -

Nelarabine) Injection • Severe Neurologic Reactions Have Been Reported
HIGHLIGHTS OF PRESCRIBING INFORMATION --------------------- DOSAGE FORMS AND STRENGTHS -------------- These highlights do not include all the information needed to use 250 mg/50 mL (5 mg/mL) vial (3) ARRANON safely and effectively. See full prescribing information for -------------------------------CONTRAINDICATIONS------------------------ ARRANON. None. ----------------------- WARNINGS AND PRECAUTIONS ---------------- ARRANON (nelarabine) Injection • Severe neurologic reactions have been reported. Monitor for signs and Initial U.S. Approval: 2005 symptoms of neurologic toxicity. (5.1) WARNING: NEUROLOGIC ADVERSE REACTIONS • Hematologic Reactions: Complete blood counts including platelets should See full prescribing information for complete boxed warning. be monitored regularly. (5.2) Severe neurologic adverse reactions have been reported with the use of • Fetal harm can occur if administered to a pregnant woman. Women should ARRANON. These adverse reactions have included altered mental states be advised not to become pregnant when taking ARRANON. (5.3) including severe somnolence, central nervous system effects including ------------------------------ ADVERSE REACTIONS ----------------------- convulsions, and peripheral neuropathy ranging from numbness and The most common (≥ 20%) adverse reactions were: paresthesias to motor weakness and paralysis. There have also been • Adult: anemia, thrombocytopenia, neutropenia, nausea, diarrhea, reports of adverse reactions associated with demyelination, and ascending vomiting, constipation, fatigue, -

The Effect of Chloramphenicol on BB88 Murine Erythroleukemia Cells
Western Michigan University ScholarWorks at WMU Dissertations Graduate College 8-2007 The Effect of Chloramphenicol on BB88 Murine Erythroleukemia Cells Peter K. W. Harris Western Michigan University Follow this and additional works at: https://scholarworks.wmich.edu/dissertations Part of the Chemistry Commons Recommended Citation Harris, Peter K. W., "The Effect of Chloramphenicol on BB88 Murine Erythroleukemia Cells" (2007). Dissertations. 872. https://scholarworks.wmich.edu/dissertations/872 This Dissertation-Open Access is brought to you for free and open access by the Graduate College at ScholarWorks at WMU. It has been accepted for inclusion in Dissertations by an authorized administrator of ScholarWorks at WMU. For more information, please contact [email protected]. THE EFFECT OF CHLORAMPHENICOL ON BB88 MURINE ERYTHROLEUKEMIA CELLS by Peter K. W. Harris A Dissertation Submitted to the Faculty o f The Graduate College in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Department of Biological Sciences Western Michigan University Kalamazoo, Michigan August 2007 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. THE EFFECT OF CHLORAMPHENICOL ON BB88 MURINE ERYTHROLEUKEMIA CELLS Peter K. W. Harris, Ph.D. Western Michigan University, 2007 DNA microarrays can be used to measure genome-wide transcript levels. These measurements may be useful in understanding cellular changes induced by a chemical agent. In this study, Affymetrix microarray technology has been used to study the effects of chloramphenicol, an antibiotic that inhibits bacterial and mitochondrial protein synthesis, on the transcription profile in mammalian cells. Transcript levels in BB88 murine erythroleukemia cells treated with 50 micromolar (pM) chloramphenicol, a concentration shown to inhibit BB 88 proliferation, are measured. -

Identification and Analysis of Single-Nucleotide Polymorphisms in the Gemcitabine Pharmacologic Pathway
The Pharmacogenomics Journal (2004) 4, 307–314 & 2004 Nature Publishing Group All rights reserved 1470-269X/04 $30.00 www.nature.com/tpj ORIGINAL ARTICLE Identification and analysis of single-nucleotide polymorphisms in the gemcitabine pharmacologic pathway AK Fukunaga1 ABSTRACT 2 Significant variability in the antitumor efficacy and systemic toxicity of S Marsh gemcitabine has been observed in cancer patients. However, there are 1 DJ Murry currently no tools for prospective identification of patients at risk for TD Hurley3 untoward events. This study has identified and validated single-nucleotide HL McLeod2 polymorphisms (SNP) in genes involved in gemcitabine metabolism and transport. Database mining was conducted to identify SNPs in 14 genes 1Department of Clinical Pharmacy and Pharmacy involved in gemcitabine metabolism. Pyrosequencing was utilized to Practice, Purdue University, W. Lafayette, IN, determine the SNP allele frequencies in genomic DNA from European and 2 USA; Departments of Medicine, Genetics, and African populations (n ¼ 190). A total of 14 genetic variants (including 12 Molecular Biology and Pharmacology, Washington University School of Medicine and SNPs) were identified in eight of the gemcitabine metabolic pathway genes. the Siteman Cancer Center, St Louis, MO, USA; The majority of the database variants were observed in population samples. 3Department of Biochemistry and Molecular Nine of the 14 (64%) polymorphisms analyzed have allele frequencies that Biology, Indiana University School of Medicine, were found to be significantly different between the European and African Indianapolis, IN, USA populations (Po0.05). This study provides the first step to identify markers Correspondence: for predicting variability in gemcitabine response and toxicity. Dr HL McLeod, Washington University The Pharmacogenomics Journal (2004) 4, 307–314.