Proquest Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Heterocycles 2 Daniel Palleros
Heterocycles 2 Daniel Palleros Heterocycles 1. Structures 2. Aromaticity and Basicity 2.1 Pyrrole 2.2 Imidazole 2.3 Pyridine 2.4 Pyrimidine 2.5 Purine 3. Π-excessive and Π-deficient Heterocycles 4. Electrophilic Aromatic Substitution 5. Oxidation-Reduction 6. DNA and RNA Bases 7. Tautomers 8. H-bond Formation 9. Absorption of UV Radiation 10. Reactions and Mutations Heterocycles 3 Daniel Palleros Heterocycles Heterocycles are cyclic compounds in which one or more atoms of the ring are heteroatoms: O, N, S, P, etc. They are present in many biologically important molecules such as amino acids, nucleic acids and hormones. They are also indispensable components of pharmaceuticals and therapeutic drugs. Caffeine, sildenafil (the active ingredient in Viagra), acyclovir (an antiviral agent), clopidogrel (an antiplatelet agent) and nicotine, they all have heterocyclic systems. O CH3 N HN O O N O CH 3 N H3C N N HN N OH O S O H N N N 2 N O N N O CH3 N CH3 caffeine sildenafil acyclovir Cl S N CH3 N N H COOCH3 nicotine (S)-clopidogrel Here we will discuss the chemistry of this important group of compounds beginning with the simplest rings and continuing to more complex systems such as those present in nucleic acids. Heterocycles 4 Daniel Palleros 1. Structures Some of the most important heterocycles are shown below. Note that they have five or six-membered rings such as pyrrole and pyridine or polycyclic ring systems such as quinoline and purine. Imidazole, pyrimidine and purine play a very important role in the chemistry of nucleic acids and are highlighted. -

Aminoketone, Oxazole and Thiazole Synthesis. Part 15. 2
Issue in Honor of Prof. C. D. Nenitzescu ARKIVOC 2002 (ii) 64-72 Aminoketone, oxazole and thiazole synthesis. Part 15.1 2-[4-(4-Halobenzenesulphonyl)-phenyl]-5-aryloxazoles Iosif Schiketanz,a Constantin Draghici,b Ioana Saramet,c and Alexandru T. Balaban a,d a Polytechnic University Bucharest, Organic Chemistry Department, Splaiul Independentei 313, Bucharest, Roumania b “C. D. Nenitzescu” Institute of Organic Chemistry, Roumanian Academy, Splaiul Independentei 202B, Bucharest, Roumania c Faculty of Pharmacy, Organic Chemistry Department, Traian Vuia Street 6, Bucharest, Roumania d Texas A&M University at Galveston, Department of Marine Sciences, 5007 Avenue U, Galveston, TX 77551, USA E-mail: [email protected] (received 14 Sep 2001; accepted 21 Mar 2002; published on the web 29 Mar 2002) Abstract Acylaminoacylation of aromatic hydrocarbons (benzene, toluene, meta-xylene, mesitylene) with 2-[4-benzenesulfonyl-(4-halophenyl)]-5-oxazolones in the presence of anhydrous aluminum chloride leads to 2-aza-1,4-diones 5 which cyclize under the action of phosphorus oxychloride yielding the corresponding 2-[4-(4-halobenzenesulphonyl)-phenyl]-5-aryloxazoles 6. The para- halogens are chloro or bromo atoms. Electronic absorption, vibrational, 1H-NMR and 13C-NMR spectral data are presented. The UV and NMR spectra provide evidence for the non-coplanarity of the oxazole and mesityl rings. Keywords: Amoinoketone, oxazole, thiazole, acylaminoacylation, synthesis Introduction In continuation of the previous part in this series,1 we now report the preparation -

One Hundred Years of Benzotropone Chemistry
One hundred years of benzotropone chemistry Arif Dastan*1, Haydar Kilic2,3 and Nurullah Saracoglu*1 Review Open Access Address: Beilstein J. Org. Chem. 2018, 14, 1120–1180. 1Department of Chemistry, Science Faculty, Atatürk University, doi:10.3762/bjoc.14.98 25240, Erzurum, Turkey, 2Oltu Vocational Training School, Atatürk University, 25400, Erzurum, Turkey and 3East Anotolia High Received: 27 December 2017 Technology Application and Research Center, Atatürk University, Accepted: 20 April 2018 25240, Erzurum, Turkey Published: 23 May 2018 Email: Associate Editor: B. Stoltz Arif Dastan* - [email protected]; Nurullah Saracoglu* - [email protected] © 2018 Dastan et al.; licensee Beilstein-Institut. License and terms: see end of document. * Corresponding author Keywords: benzotropolone; benzotropone; dibenzotropone; halobenzotropolone; halobenzotropone; tribenzotropone; tropone Abstract This review focuses on the chemistry of benzo-annulated tropones and tropolones reported since the beginning of the 20th century, which are currently used as tools by the synthetic and biological communities. Review 1. Introduction Tropone (1) and tropolone (2) have fascinated organic chemists have been reported in the literature in a number of natural forms for well over one hundred years. The carbocycles 1 and 2 are a (Figure 1) [1-8]. These compounds have a structural class special variety of organic compounds and represent a nonben- spacing from the simple monocyclic tropones, such as the po- zenoid type of aromatic system (Scheme 1). Their dipolar reso- tent antifungal and antibiotic monoterpene β-thujaplicin (4) nance structures such as tropylium oxide form 1B and 2B have [17-23] (isolated from the heartwood and essential oils of trees been reported to provide a Hückel sextet of electrons that is of the family Cupressaceae), to complex macrocyclic ana- necessary for aromaticity (Scheme 1) [1-9]. -

In Vitroo and in Vivo Pharmacology of 4-Substituted
IN VITROO AND IN VIVO PHARMACOLOGY OF 4-SUBSTITUTED METHOXYBENZOYL-ARYL-THIAZOLES (SMART) AND 2- ARYLTHIAZOLIDINE-4-CARBOXYLIC ACID AMIDES (ATCAA) Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Chien-Ming Li, M.S. Graduate Program in Pharmacy The Ohio State University 2010 Dissertation Committee: Dr. James T. Dalton, Advisor Dr. Robert W. Brueggemeier Dr. Thomas D. Schmittgen Dr. Mitch A. Phelps Copyright by Chien-Ming Li 2010 ABSTRACT Formation of microtubules is a dynamic process that involves polymerization and depolymerization of the αβ-tubulin heterodimers. Drugs that enhance or inhibit tubulin polymerization can destroy this dynamic process, arresting cells in the G2/M phase of the cell cycle. Although drugs that target tubulin generally demonstrate cytotoxic potency in sub-nanomolar range, resistance due to drug efflux is a common phenomenon among the antitubulin agents. We recently reported a class of 4-Substituted Methoxybenzoyl-Aryl- Thiazoles (SMART), which exhibited great in vitro and in vivo potency. SMART compounds effectively inhibited tubulin polymerization in dose dependent manner, suggesting that SMART compounds may bind to tubulin and subsequently hamper the polymerization. To date the only method to determine the binding of inhibitor to tubulin has been competitive radioligand binding assays. We developed a novel non-radioactive mass spectrometry (MS) binding assay to study the tubulin binding of colchicine, vinblastine and paclitaxel. The method involves a very simple step of separating the unbound ligand from macromolecules using ultrafiltration. The unbound ligand in the filtrate can be accurately determined using a highly sensitive and specific LC-MS/MS method. -

Heterocyclic Chemistrychemistry
HeterocyclicHeterocyclic ChemistryChemistry Professor J. Stephen Clark Room C4-04 Email: [email protected] 2011 –2012 1 http://www.chem.gla.ac.uk/staff/stephenc/UndergraduateTeaching.html Recommended Reading • Heterocyclic Chemistry – J. A. Joule, K. Mills and G. F. Smith • Heterocyclic Chemistry (Oxford Primer Series) – T. Gilchrist • Aromatic Heterocyclic Chemistry – D. T. Davies 2 Course Summary Introduction • Definition of terms and classification of heterocycles • Functional group chemistry: imines, enamines, acetals, enols, and sulfur-containing groups Intermediates used for the construction of aromatic heterocycles • Synthesis of aromatic heterocycles • Carbon–heteroatom bond formation and choice of oxidation state • Examples of commonly used strategies for heterocycle synthesis Pyridines • General properties, electronic structure • Synthesis of pyridines • Electrophilic substitution of pyridines • Nucleophilic substitution of pyridines • Metallation of pyridines Pyridine derivatives • Structure and reactivity of oxy-pyridines, alkyl pyridines, pyridinium salts, and pyridine N-oxides Quinolines and isoquinolines • General properties and reactivity compared to pyridine • Electrophilic and nucleophilic substitution quinolines and isoquinolines 3 • General methods used for the synthesis of quinolines and isoquinolines Course Summary (cont) Five-membered aromatic heterocycles • General properties, structure and reactivity of pyrroles, furans and thiophenes • Methods and strategies for the synthesis of five-membered heteroaromatics -

Recent Advances in the Synthesis of Oxazole-Based Molecules Via Van Leusen Oxazole Synthesis
molecules Review Recent Advances in the Synthesis of Oxazole-Based Molecules via van Leusen Oxazole Synthesis Xunan Zheng 1,2, Wei Liu 3,* and Dawei Zhang 1,* 1 College of Chemistry, Jilin University, Changchun 130012, China; [email protected] 2 College of Plant Science, Jilin University, Changchun 130062, China 3 Department of Pesticide Science, Plant Protection College, Shenyang Agricultural University, Shenyang 110866, China * Correspondence: [email protected] (W.L.); [email protected] (D.Z.); Tel.: +86-188-1775-2588 (W.L.); +86-431-8783-6471 (D.Z.) Academic Editors: Anna Carbone and Fabio Bertozzi Received: 2 March 2020; Accepted: 23 March 2020; Published: 31 March 2020 Abstract: Oxazole compounds, including one nitrogen atom and one oxygen atom in a five-membered heterocyclic ring, are present in various biological activities. Due to binding with a widespread spectrum of receptors and enzymes easily in biological systems through various non-covalent interactions, oxazole-based molecules are becoming a kind of significant heterocyclic nucleus, which have received attention from researchers globally, leading them to synthesize diverse oxazole derivatives. The van Leusen reaction, based on tosylmethylisocyanides (TosMICs), is one of the most appropriate strategies to prepare oxazole-based medicinal compounds. In this review, we summarize the recent advances of the synthesis of oxazole-containing molecules utilizing the van Leusen oxazole synthesis from 1972, aiming to look for potential oxazole-based medicinal compounds, which are valuable information for drug discovery and synthesis. Keywords: van Leusen; TosMICs; oxazole; synthesis 1. Introduction The oxazole ring, with one nitrogen atom and one oxygen atom, which are widely displayed in natural products and synthetic molecules, is known as a prime skeleton for drug discovery. -

Total Synthesis of Phorbazole B
molecules Article Total Synthesis of Phorbazole B Yngve Guttormsen 1 , Magnus E. Fairhurst 2, Sunil K. Pandey 2, Johan Isaksson 1 , Article Bengt Erik Haug 2,* and Annette Bayer 1,* Total Synthesis of Phorbazole B 1 Department of Chemistry, UiT The Arctic University of Norway, Hansine Hansens veg 54, 9037 Tromsø, Norway; [email protected] (Y.G.); [email protected] (J.I.) Yngve Guttormsen 1, Magnus E. Fairhurst 2, Sunil K. Pandey 2, Johan Isaksson 1, 2 DepartmentBengt Erik of Haug Chemistry 2,* and andAnnette Centre Bayer for Pharmacy,1,* University of Bergen, Allégaten 41, 5007 Bergen, Norway; [email protected] (M.E.F.); [email protected] (S.K.P.) 1 * Correspondence: Department of [email protected] Chemistry, UiT The Arctic (B.E.H.);University [email protected] of Norway, Hansine Hansens (A.B.); veg 54, Tel.: +47-55-58-34-689037 Tromsø, Norway; (B.E.H.); [email protected]+47-77-64-40-69 (A.B.) (Y.G.); [email protected] (J.I.) 2 Department of Chemistry and Centre for Pharmacy, University of Bergen, Allégaten 41, Academic Editors:5007 Bergen, Magne Norway; Olav [email protected] Sydnes and Joanne Harvey(M.E.F.); [email protected] (S.K.P.) Received:* 19Correspondence: September 2020; bengt Accepted:[email protected] 18 October (B.E.H.); 2020; [email protected] Published: 21 October(A.B.); 2020 Tel.:+47-55-58-34-68 (B.E.H.); +47-77-64-40-69 (A.B.) Abstract:AcademicPhorbazoles Editors: Magne are polychlorinated Olav Sydnes and Joanne heterocyclic Harvey secondary metabolites isolated from a marine spongeReceived: and several 19 September of these 2020 natural; Accepted: products 18 October have2020; Published: shown inhibitory21 October 2020 activity against cancer cells. -

Ammonia Synthons for the Multicomponent Assembly of Complex Γ-Lactams
Ammonia synthons for the multicomponent SPECIAL FEATURE assembly of complex γ-lactams Darlene Q. Tan, Kevin S. Martin, James C. Fettinger, and Jared T. Shaw1 Department of Chemistry, University of California, One Shields Avenue, Davis, CA 95616 Edited by Stuart L. Schreiber, Broad Institute, Cambridge, MA, and approved December 21, 2010 (received for review November 8, 2010) The synthesis of γ-lactams that are unsubstituted at the 1-position (nitrogen) as well as their subsequent N-functionalization is reported. A recently discovered four-component reaction (4CR) is employed with either an ammonia precursor or a protected form of ammonia that can be deprotected in a subsequent synthetic step. These methods represent the first multicomponent assembly of complex lactam structures that are unsubstituted at nitrogen. In addition, two methods for the introduction of nitrogen substitu- ents that are not possible through the original 4CR are reported. X-ray crystallographic analysis of representative structures reveals conformational changes in the core structure that will enable future deployment of this chemistry in the design and synthesis of diverse collections of lactams suitable for the discovery of new biological probes. multicomponent reaction ∣ stereoselective CHEMISTRY ive-membered ring lactams, which are known as γ-lactams or F2-oxopyrrolidines, are important structural motifs in biologi- cally active natural products that are also found in medicinal leads and approved drugs (Fig. 1). Heliotropamide (1, 2) and bisavenanthramide B (3, 4) are examples of ferrulic acid amides that undergo biosynthetic dimerization to produce γ-lactams, whereas lactacystin (5) and salinosporamide (6) emanate from more complex biosyntheses. Although these compounds share the γ-lactam core, an important difference emerges in the substi- Fig. -

Essentials of Heterocyclic Chemistry-I Heterocyclic Chemistry
Baran, Richter Essentials of Heterocyclic Chemistry-I Heterocyclic Chemistry 5 4 Deprotonation of N–H, Deprotonation of C–H, Deprotonation of Conjugate Acid 3 4 3 4 5 4 3 5 6 6 3 3 4 6 2 2 N 4 4 3 4 3 4 3 3 5 5 2 3 5 4 N HN 5 2 N N 7 2 7 N N 5 2 5 2 7 2 2 1 1 N NH H H 8 1 8 N 6 4 N 5 1 2 6 3 4 N 1 6 3 1 8 N 2-Pyrazoline Pyrazolidine H N 9 1 1 5 N 1 Quinazoline N 7 7 H Cinnoline 1 Pyrrolidine H 2 5 2 5 4 5 4 4 Isoindole 3H-Indole 6 Pyrazole N 3 4 Pyrimidine N pK : 11.3,44 Carbazole N 1 6 6 3 N 3 5 1 a N N 3 5 H 4 7 H pKa: 19.8, 35.9 N N pKa: 1.3 pKa: 19.9 8 3 Pyrrole 1 5 7 2 7 N 2 3 4 3 4 3 4 7 Indole 2 N 6 2 6 2 N N pK : 23.0, 39.5 2 8 1 8 1 N N a 6 pKa: 21.0, 38.1 1 1 2 5 2 5 2 5 6 N N 1 4 Pteridine 4 4 7 Phthalazine 1,2,4-Triazine 1,3,5-Triazine N 1 N 1 N 1 5 3 H N H H 3 5 pK : <0 pK : <0 3 5 Indoline H a a 3-Pyrroline 2H-Pyrrole 2-Pyrroline Indolizine 4 5 4 4 pKa: 4.9 2 6 N N 4 5 6 3 N 6 N 3 5 6 3 N 5 2 N 1 3 7 2 1 4 4 3 4 3 4 3 4 3 3 N 4 4 2 6 5 5 5 Pyrazine 7 2 6 Pyridazine 2 3 5 3 5 N 2 8 N 1 2 2 1 8 N 2 5 O 2 5 pKa: 0.6 H 1 1 N10 9 7 H pKa: 2.3 O 6 6 2 6 2 6 6 S Piperazine 1 O 1 O S 1 1 Quinoxaline 1H-Indazole 7 7 1 1 O1 7 Phenazine Furan Thiophene Benzofuran Isobenzofuran 2H-Pyran 4H-Pyran Benzo[b]thiophene Effects of Substitution on Pyridine Basicity: pKa: 35.6 pKa: 33.0 pKa: 33.2 pKa: 32.4 t 4 Me Bu NH2 NHAc OMe SMe Cl Ph vinyl CN NO2 CH(OH)2 4 8 5 4 9 1 3 2-position 6.0 5.8 6.9 4.1 3.3 3.6 0.7 4.5 4.8 –0.3 –2.6 3.8 6 3 3 5 7 4 8 2 3 5 2 3-position 5.7 5.9 6.1 4.5 4.9 4.4 2.8 4.8 4.8 1.4 0.6 3.8 4 2 6 7 7 3 N2 N 1 4-position -

Antimicrobial Activity and Characterization of Some Oxazole, Thiazol and Quinoline
1222 Indian Journal of Forensic Medicine & Toxicology, January-March 2020, DOIVol. 14, Number: No. 1 10.37506/v14/i1/2020/ijfmt/193076 Antimicrobial Activity And Characterization of Some Oxazole, Thiazol And Quinoline Amjad Gali. Eliwi1, Safaa. Abdul-Hameed. Dadoosh1, Zainab Z. Mohammed Ali 1, Abdul Jabar Kalaf Atia1, Iman Rajab .Mohammed 1, Zaman Ahmed. Hussein1 1Department of Chemistry, College of Science-University of AL-Mustansiriyah- Baghdad-Iraq Abstract New Heterocyclic compounds derivatives comprising 1,3-oxazole, chalcone ,thiazole, pyrimidine, quinolone moieties are reported. New derivatives of Quinazolin-4 (3H)-one ring comprising Schiff’s bases,(1,3,4- Thiadiazole),(1,3,4-Oxadiazole) and (1,2,4-Triazole), Thiaurease moieties are reported. Compounds (1), (2) and (5) were synthesized by reaction of benzoyl chloride with urea , thiourea and anthranilic acid respectively , then compounds (1)and (2) were converted into(3a-c) and (4a-c) derivatives. While compound (5) reaction with urea to convert to compound (6) which was converted to (7a-c) . chalcone derivatives (9a,b ) were readily obtained by reaction of compound (8) with different aldehydes, Compounds (9a , b) were converted into (10a,b) and (11a,b) The structure of these compounds has been established on the basis of their spectral data FTIR and 1HNMR. These compounds were tested for invitro antibacterial activity against Escherichia coli, Sepidermidis, S.aureusand Klebseillastandard methods. These synthesized compounds have been shown moderate to good antibacterial activity. Keywords: 1,3 oxazole, chalcone, thiazole, pyrimidine, quinolone, antimicrobial activity. Introduction antitubercular activities 21 and anti-inflammatory activities 22 . Many Pyrimidinederivatives are used for Heterocyclic compounds had been receiving thyroid drugs and leukemia 13 . -
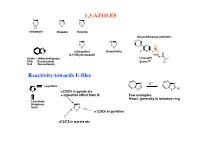
1,3-AZOLES Reactivity Towards E-Files
1,3-AZOLES N N N N S O H Imidazole Thiazole Oxazole Oxazolidineone antibiotic N HN O N O N O O 2-Oxazoline Oxazolidine F N O X (4,5-Dihydrooxazol) H N X=NH: 1H-Benzimidazole Linesolid X=O: Benzoxazole ZyvoxTM O X=S Benzotiazole Reactivity towards E-files N E+ N N ≈ pyridine X X E X ≈C3/C4 in pyrole etc + inductive effect from N Few examples React. generally in benzene ring ≈ pyrazole thiophene N furan X ≈ C2/C6 in pyridine ≈C2/C5 in pyrole etc Reaction with electrophiles on N - Protonation H H pKa H N H+ N N N H X=NH: 7.1 N X X X=S: 2.5 N N X=O: 0.8 H H O stabilized Destabilized R R N HN Taut.: N N H R= Me: ca 1 : 1 R=NO2: ca 400 : 1 Reaction with electrophiles on N R 'R R 'R R N-Alkylation N N N N N N H H R'-X R N R-X N - H+ R R R X X HN HN N May be sterically favoured Reactivity: N N N R' R' X = N-Me X=S X=O 900 : 15 : 1 R R R N N N N low react. N N N N H R' SO2Ph Base R'-X - H+ R R 'R R HN N N N N N N N H Base Et Et N + - N N PhCOCl 1) Et3O BF4 N N N O O HN N steric control Reaction with electrophiles on N N-Acylation Only rel. -

Rsc Cc C3cc48467j 1..6
1 ChemComm 1 5 COMMUNICATION 5 Converting oxazoles into imidazoles: new Q1 Q2 10 opportunities for diversity-oriented synthesis† 10 Cite this: DOI: 10.1039/c3cc48467j Thibaut Alzieu,a Johannes Lehmann,a Ajay B. Naidu,b Rainer E. Martin*a and Received 5th November 2013, Robert Britton*b Accepted 20th December 2013 15 DOI: 10.1039/c3cc48467j 15 www.rsc.org/chemcomm We report the optimization of a neglected reaction for the rapid and 20 direct conversion of oxazoles into N-substituted imidazoles. The 20 utility of this microwave-promoted reaction for diversity oriented synthesis is demonstrated in the preparation of >40 N-substituted imidazoles, including a-imidazolyl esters. 25 Imidazoles are essential heterocycles in modern medicinal 25 chemistry1 andcommoninpharmaceuticals2 and natural products.3 As such, tremendous effort has been devoted to both imidazole synthesis4 and functionalization,5 from which many robust pro- cesses have emerged.6 For example, the van Leusen imidazole 30 synthesis7 or multi-component reactions of 1,2-diketones with alde- 30 hydes and amines8 are standard protocols. Despite these advances, several fundamental challenges to imidazole synthesis include the regioselective N-alkylation of asymmetric imidazoles,9 production of N-tertiary alkyl imidazoles,10 andtherapidsynthesisofdiverse 35 libraries of N-substituted imidazoles.11 Considering the ease with 35 which oxazoles are prepared12,13 and the well-established processes for their selective functionalization at C214 or C5,15 they represent potentially useful synthetic isostereos for imidazole. Thus, a direct conversion of oxazoles into imidazoles would provide unique Scheme 1 Cornforth’s imidazole synthesis and related reaction of phenyl 40 opportunities for the regioselective synthesis of N-functionalized 40 oxazole.