MULTAQ, INN-Dronedarone
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
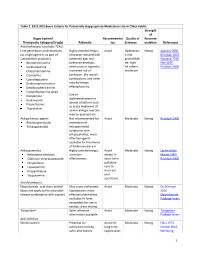
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Safety and Efficacy of Dronedarone from Clinical Trials to Real
Europace (2019) 21, 1764–1775 REVIEW doi:10.1093/europace/euz193 Safety and efficacy of dronedarone from clinical Downloaded from https://academic.oup.com/europace/article-abstract/21/12/1764/5536329 by Uppsala Universitetsbibliotek user on 20 February 2020 trials to real-world evidence: implications for its use in atrial fibrillation Giuseppe Boriani1, Carina Blomstro¨m-Lundqvist2, Stefan H. Hohnloser3, Lennart Bergfeldt4,5, Giovanni L. Botto6, Alessandro Capucci7, Ignacio Ferna´ndez Lozano8, Andreas Goette9,10, Carsten W. Israel3,11, Jose´ L. Merino12, and A. John Camm13* 1Division of Cardiology, Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Policlinico di Modena, Modena, Italy; 2Department of Medical Science and Cardiology, Uppsala University, Uppsala, Sweden; 3Division of Clinical Electrophysiology, Department of Cardiology, J W Goethe University, Frankfurt, Germany; 4Department of Molecular and Clinical Medicine, Institute of Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden; 5Va¨stra Go¨taland, Department of Cardiology, Sahlgrenska University Hospital, Gothenburg, Sweden; 6ASST Rhodense, Ospedale di Circolo Rho, Milan, Italy; 7Universita` Politecnica delle Marche, Ancona, Italy; 8Sociedad Espanola~ de Cardiologı´a, Madrid, Spain; 9Medical Clinic II, Cardiology Department, St Vincenz-Krankenhaus Paderborn, Paderborn, Germany; 10Working Group Molecular Electrophysiology, University Hospital Magdeburg, Magdeburg, Germany; 11Clinic of Internal Medicine, Bethel-Clinic, Bielefeld, Germany; 12Arrhythmia & Robotic EP Unit, Hospital Universitario La Paz-IdiPaz, Madrid, Spain; and 13Cardiology Clinical Academic Group, Molecular and Clinical Sciences Institute, St George’s University of London, Cranmer Terrace, London SW17 0RE, UK Received 17 May 2019; editorial decision 13 June 2019; accepted 20 June 2019; online publish-ahead-of-print 19 July 2019 Efficacy and safety of dronedarone was shown in the ATHENA trial for paroxysmal or persistent atrial fibrillation (AF) patients. -

Medication Guide MULTAQ (MUL-Tak) (Dronedarone) Tablets
17.2 Medication Guide Medication Guide MULTAQ (MUL-tak) (dronedarone) Tablets Read this Medication Guide before you start taking MULTAQ and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment. What is the most important information I should know about MULTAQ? MULTAQ can cause serious side effects. Do not take MULTAQ if you: 1. have symptoms of heart failure that recently worsened and you were hospitalized, or if you have severe heart failure. MULTAQ doubles your risk of dying if you have these conditions. Heart failure means your heart does not pump blood through your body as well as it should. Call your doctor right away if you have any signs or symptoms of heart failure during treatment with MULTAQ: • shortness of breath or wheezing at rest • wheezing, chest tightness or coughing up frothy sputum at rest, nighttime or after minor exercise • trouble sleeping or waking up at night because of breathing problems • using more pillows to prop yourself up at night so you can breathe more easily • gaining more than 5 pounds quickly • increasing swelling of feet or legs 2. have a type of atrial fibrillation (irregular heart rhythm) called permanent atrial fibrillation (AF). You and your doctor may decide not to try to change your heart rhythm back to a normal heart rhythm or your heart rhythm cannot be changed back to a normal rhythm. If you have permanent AF and take MULTAQ, you have a higher risk of death, stroke, and needing to be treated in a hospital for your heart failure. -

Multaq, INN-Dronedarone
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT MULTAQ 400 mg film-coated tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each tablet contains 400 mg of dronedarone (as hydrochloride). Excipient with known effect: Each tablet also contains 41.65 mg of lactose (as monohydrate). For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Film-coated tablet (tablet). White, oblong shaped tablets, engraved with a double wave marking on one side and “4142”code on the other side. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications MULTAQ is indicated for the maintenance of sinus rhythm after successful cardioversion in adult clinically stable patients with paroxysmal or persistent atrial fibrillation (AF). Due to its safety profile (see sections 4.3 and 4.4), MULTAQ should only be prescribed after alternative treatment options have been considered. MULTAQ must not be given to patients with left ventricular systolic dysfunction or to patients with current or previous episodes of heart failure. 4.2 Posology and method of administration Treatment should be initiated and monitored only under specialist supervision (see section 4.4). Treatment with dronedarone can be initiated in an outpatient setting. Treatment with Class I or III antiarrhythmics (such as flecainide, propafenone, quinidine, disopyramide, dofetilide, sotalol, amiodarone) must be stopped before starting dronedarone. There is limited information on the optimal timing to switch from amiodarone to dronedarone. It should be considered that amiodarone may have a long duration of action after discontinuation due to its long half-life. If a switch is envisaged, this should be done under the supervision of a specialist (see sections 4.3 and 5.1). -

Guideline for Preoperative Medication Management
Guideline: Preoperative Medication Management Guideline for Preoperative Medication Management Purpose of Guideline: To provide guidance to physicians, advanced practice providers (APPs), pharmacists, and nurses regarding medication management in the preoperative setting. Background: Appropriate perioperative medication management is essential to ensure positive surgical outcomes and prevent medication misadventures.1 Results from a prospective analysis of 1,025 patients admitted to a general surgical unit concluded that patients on at least one medication for a chronic disease are 2.7 times more likely to experience surgical complications compared with those not taking any medications. As the aging population requires more medication use and the availability of various nonprescription medications continues to increase, so does the risk of polypharmacy and the need for perioperative medication guidance.2 There are no well-designed trials to support evidence-based recommendations for perioperative medication management; however, general principles and best practice approaches are available. General considerations for perioperative medication management include a thorough medication history, understanding of the medication pharmacokinetics and potential for withdrawal symptoms, understanding the risks associated with the surgical procedure and the risks of medication discontinuation based on the intended indication. Clinical judgement must be exercised, especially if medication pharmacokinetics are not predictable or there are significant risks associated with inappropriate medication withdrawal (eg, tolerance) or continuation (eg, postsurgical infection).2 Clinical Assessment: Prior to instructing the patient on preoperative medication management, completion of a thorough medication history is recommended – including all information on prescription medications, over-the-counter medications, “as needed” medications, vitamins, supplements, and herbal medications. Allergies should also be verified and documented. -

Ventricular Tachycardia Drugs Versus Devices John Camm St
Cardiology Update 2015 Davos, Switzerland: 8-12th February 2015 Ventricular Arrhythmias Ventricular Tachycardia Drugs versus Devices John Camm St. George’s University of London, UK Imperial College, London, UK Declaration of Interests Chairman: NICE Guidelines on AF, 2006; ESC Guidelines on Atrial Fibrillation, 2010 and Update, 2012; ACC/AHA/ESC Guidelines on VAs and SCD; 2006; NICE Guidelines on ACS and NSTEMI, 2012; NICE Guidelines on heart failure, 2008; NICE Guidelines on Atrial Fibrillation, 2006; ESC VA and SCD Guidelines, 2015 Steering Committees: multiple trials including novel anticoagulants DSMBs: multiple trials including BEAUTIFUL, SHIFT, SIGNIFY, AVERROES, CASTLE- AF, STAR-AF II, INOVATE, and others Events Committees: one trial of novel oral anticoagulants and multiple trials of miscellaneous agents with CV adverse effects Editorial Role: Editor-in-Chief, EP-Europace and Clinical Cardiology; Editor, European Textbook of Cardiology, European Heart Journal, Electrophysiology of the Heart, and Evidence Based Cardiology Consultant/Advisor/Speaker: Astellas, Astra Zeneca, ChanRX, Gilead, Merck, Menarini, Otsuka, Sanofi, Servier, Xention, Bayer, Boehringer Ingelheim, Bristol- Myers Squibb, Daiichi Sankyo, Pfizer, Boston Scientific, Biotronik, Medtronic, St. Jude Medical, Actelion, GlaxoSmithKline, InfoBionic, Incarda, Johnson and Johnson, Mitsubishi, Novartis, Takeda Therapy for Ventricular Tachycardia Medical therapy Antiarrhythmic drugs Autonomic management Ventricular tachycardia Monomorphic Polymorphic Ventricular fibrillation Ventricular storms Ablation therapy Device therapy Surgical Defibrillation Catheter Antitachycardia pacing History of Antiarrhythmic Drugs 1914 - Quinidine 1950 - Lidocaine 1951 - Procainamide 1946 – Digitalis 1956 – Ajmaline 1962 - Verapamil 1962 – Disopyramide 1964 - Propranolol 1967 – Amiodarone 1965 – Bretylium 1972 – Mexiletine 1973 – Aprindine, Tocainide 1969 - Diltiazem 1975- Flecainide 1976 – Propafenone Encainide Ethmozine 2000 - Sotalol D-sotalol 1995 - Ibutilide (US) Recainam 2000 – Dofetilide US) IndecainideX Etc. -

MULTAQ (Dronedarone) Tablets Suspension Or Discontinuation of MULTAQ (5.1) Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION • Pregnancy (4, 8.1) These highlights do not include all the information needed to use • Nursing mothers (4, 8.3) MULTAQ safely and effectively. See full prescribing information for MULTAQ. -----------------------WARNINGS AND PRECAUTIONS------------------------ • Heart failure: If heart failure develops or worsens, consider the MULTAQ (dronedarone) Tablets suspension or discontinuation of MULTAQ (5.1) Initial U.S. Approval: 2009 • Hypokalemia and hypomagnesemia: Maintain potassium and magnesium levels within the normal range (5.2) WARNING: HEART FAILURE • QT prolongation: Stop MULTAQ if QTc Bazett ≥500ms (5.3) MULTAQ is contraindicated in patients with NYHA Class IV heart • Increase in creatinine: Within a week, MULTAQ causes a small increase failure or NYHA Class II - III heart failure with a recent in serum creatinine that does not reflect a change in underlying renal decompensation requiring hospitalization or referral to a specialized function (5.4) heart failure clinic (4). • Teratogen: Women of childbearing potential should use effective contraception while using MULTAQ (5.5) In a placebo-controlled study in patients with severe heart failure requiring recent hospitalization or referral to a specialized heart failure ------------------------------ADVERSE REACTIONS------------------------------- clinic for worsening symptoms (the ANDROMEDA Study), patients Most common adverse reactions (≥2%) are diarrhea, nausea, abdominal pain, given dronedarone had a greater than two-fold increase in mortality. -

Australian Public Assessment Report for Dronedarone Hydrochloride
Australian Public Assessment Report for Dronedarone Hydrochloride Proprietary Product Name: Multaq, Dronedarone Winthrop, Dronedarone Sanofi Submission No: PM-2008-3045-3 Sponsor: Sanofi Aventis Australia Pty Ltd October 2010 Contents I. Introduction to Product Submission........................................................................3 Submission Details.................................................................................................................. 3 Product Background................................................................................................................ 3 Regulatory Status .................................................................................................................... 4 Product Information ................................................................................................................ 4 II. Quality Findings.........................................................................................................4 Drug Substance (active ingredient)......................................................................................... 4 Drug Product........................................................................................................................... 5 Bioavailability......................................................................................................................... 5 Quality Summary and Conclusions......................................................................................... 7 III. Nonclinical Findings -

PHARMACY TIMES by IEHP PHARMACEUTICAL SERVICES DEPARTMENT February 11, 2013
PHARMACY TIMES BY IEHP PHARMACEUTICAL SERVICES DEPARTMENT February 11, 2013 The Centers for Medicare and Medicaid Services (CMS) developed performance and quality measures to help Medicare beneficiaries make informed decisions regarding health and prescription drug plans. As part of this effort, CMS adopted measures for High Risk Medication (HRM) endorsed by the Pharmacy Quality Alliance (PQA) and the National Quality Forum (NQF). The HRM was developed using existing HEDIS measurement “Drugs to be avoided in the elderly”. The HRM rate analyzes the percentage of Medicare Part D beneficiaries 65 years or older who have received prescriptions for drugs with a high risk of serious side effects in the elderly. In order to advance patient safety, IEHP will be identifying members over 65 and currently on one of the medications identified in Table 1. Providers will be receiving a list of these members from IEHP on an ongoing basis. IEHP asks providers to review their member’s current drug regimen and safety risk and make any appropriate changes when applicable. Table 1: Medications identified by CMS to be high risk in the elderly: Drug Class Drug Safety Concerns IEHP Formulary Alternative(s) Acetylcholinesterase Donepezil (in patients Orthostatic hypotension or Memantine Inhibitor with syncope) bradycardia Amphetamines Dextroamphetamine CNS stimulation Weight Control: Diet Lisdextroamphetamine & lifestyle Diethylpropion modification Methylphenidate Phentermine Depression: mirtazapine, trazodone Analgesic Pentazocine Confusion, hallucination, Mild Pain: (includes Meperidine delirium, fall, fracture APAP combination Tramadol Lowers seizure threshold medications) Aspirin > 325 mg/day GI bleeding/peptic ulcer, edema Mod-Severe Pain Diflunisal may worsen heart failure Norco Etodolac Vicodin Fenoprofen Percocet Ketoprofen Morphine Meclofenamate Mefenamic acid Nabumetone 303 E. -

Dronedarone (Multaq ®)
Dronedarone (Multaq ®) What is dronedarone? What if you miss taking a dose? Dronedarone is a medication that If it is almost time for you next dose, skip helps the heart beat regularly when you the dose you missed and just take your next have had abnormal heartbeats. scheduled dose. It is a member of a drug class called Never take two doses at the same time. anti-arrhythmics. Anti-arrhythmics help If you have questions about taking your control the disorganized electrical medications, or missed doses, contact your impulses that lead to irregular heart pharmacist or your health care provider. In rhythms. British Columbia, you can also call 8-1-1. What is dronedarone used for? Do not take any of the following without . Atrial Fibrillation or Atrial Flutter checking with your health care provider . Other irregular heart rhythms . Cough or cold medicines . St. John’s Wort . Herbal products . Chinese medicines How does dronedarone help with . Grapefruit/grapefruit juice abnormal heartbeats? . Other medicines you can buy at a . Reduces episodes of irregular pharmacy rhythm . Reduces length of time in Health Provider contact information: irregular rhythm . Improves patient symptoms and quality of life To learn more about dronedarone, go to the HealthLinkBC web site (www.healthlinkbc.ca) or call 8-1-1. How should I take my dronedarone? Take this medication regularly, as prescribed by your doctor. Did you know? It can take several weeks, Dronedarone doses should be spaced and sometimes months, before you notice evenly throughout the day and taken your heart rhythm improving and you with food. start feeling better. -

Increased)Mortality) With)Dronedarone:)) Is)Digoxin)The)Cause?)
Increased)mortality) with)dronedarone:)) is)digoxin)the)cause?) Alessandro Capucci, MD Department of Cardiology, Marche Polytechnic University, Ancona, Italy NO CONFLICT OF INTEREST TO DECLARE DRONEDARONE' Dronedarone'is'a'noniodinated'benzofuran' deriva6ve'related'to'amiodarone' ! " class!I"IV!an*arrythmic!ac*vity!! " an*adrenergic!effects! " an*fibrillatory!effects!on!the!atrial!and! ventricular!myocardium! " no!iodine"related!organ!toxicity,!a! decreased!lipophilicity!and!a! shortened!half"life! Kathofer et al. Cardiovasc Drug Rev.2005;23(3):217-30! Comparison'of'some'major'pharmacodynamic' proper6es'of'dronedarone'and'amiodarone'' Bramah N. Singh, and Etienne Aliot Eur Heart J Suppl 2007;9:G17-G25 EURIDIS/ADONIS' Dronedarone! Showed!a!Significant! Reduc*on! in!First!AF! Recurrence!in! Combined!Analysis! Singh BN, et al. N Engl J Med. 2007;357:987-99! POST HOC analysis of data from the EURIDIS and ADONIS trials The!aim!of!this!post!hoc!analysis!was!to!evaluate!the!efficacy!and!safety!of!dronedarone! in!pa*ents!previously!treated!with!AADs,!with!a!specific!focus!on!class!Ic!AADs!or!sotalol! The!primary!end!point!was!AF/AFL!recurrence!in!pa*ents!previously!treated! with!another!AAD!that!was!discon*nued!for!whatever!reason!prior!to! randomiza*on.! Guerra F, Capucci A et al Clin. Cardiol. 37, 12, 717–724; 2014 Results!!! In patients previously treated with any AADs, dronedarone decreased the risk of AF recurrence by 30.4% vs placebo (HR 0.70; P < 0.001) In patients previously treated with a class Ic agent, dronedarone decreased the risk of recurrence by 31.4% (HR: 0.69; P = 0.004) In patients previously treated with sotalol, dronedarone showed a trend toward a decrease of risk of recurrence (HR: 0.86; P = 0.244) Guerra F, Capucci A et al Clin. -

NHS Nottinghamshire County Black List
Medicines and Appliances of Limited Clinical Value V1.2 Last reviewed: Nov 2020 Review date: Nov 2023 Medicines and Appliances of Limited Clinical Value It is recommended that medicines classified as GREY under the Nottinghamshire Traffic Light System are not routinely prescribed on the NHS. See Notts Position Statement on Restricted Use Medicines. The grey classification is defined as ‘medicines which the Nottinghamshire Area Prescribing Committee does not recommend for use at present due to limited clinical and/or cost effective data’. In order to improve prescribing efficiency and support the release of further savings from the CCGs prescribing budget, the grey classification should be reinforced amongst all prescribers. In addition the following medicines have been agreed as having limited clinical value* and should either NEVER be prescribed on the NHS (The “NEVER” List) or only be prescribed in exceptional circumstances (The “RARELY PRESCRIBED” List). *previously known as the Low Priority List The “NEVER” List – The following products should NEVER be prescribed on the NHS in Nottinghamshire (unless agreed via the Individual Funding Request process). Product Traffic Light Comments Aliskiren (Rasilez®) GREY Not included in NICE hypertension guideline as insufficient evidence of its effectiveness. Also some safety concerns (see MHRA alert). See NHSE guidance (no exceptions) Bath and shower GREY See Notts APC Emollient formulary here. emollients Oilatum® Plus is See Nottinghamshire self care position statements RED for MRSA here. treatment at NUH where Octenisan® is Note that topical emollients / soap substitutes may still not appropriate be prescribed if self-care is not appropriate. Co-danthramer & co- GREY Risk of dantron burns and control is usually achieved danthrusate with alternative.