Identification of Novel Murine- and Human-Specific RPGRIP1 Splice Variants with Distinct Expression Profiles and Subcellular
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Characterization of the Human RPGR Proximal Promoter
Biochemistry and Molecular Biology Functional Characterization of the Human RPGR Proximal Promoter Xinhua Shu,*,1,2 Julie R. Simpson,2 Alan W. Hart,2 Zhihong Zeng,3 Sarita Rani Patnaik,1 Philippe Gautier,2 Emma Murdoch,2 Brian Tulloch,2 and Alan F. Wright*,2 PURPOSE. Mutations in the retinitis pigmentosa (RP) GTPase promoter will facilitate understanding of the functional role regulator (RPGR) gene account for more than 70% of X-linked of RPGR in the retina and gene therapy of X-linked RP. (Invest RP cases. This study aims to characterize the proximal Ophthalmol Vis Sci. 2012;53:3951–3958) DOI:10.1167/ promoter region of the human RPGR gene. iovs.11-8811 0 METHODS. The 5 -flanking region (5 kb) of human RPGR was cloned and sequenced. A potential transcription start site and etinitis pigmentosa (RP) is a genetically heterogeneous transcription factor binding motifs were identified by bio- Rgroup of retinal degenerations that affect 1 in 4000 in the informatic analysis. Constructs containing the putative human general population.1,2 Most cases are inherited in an autosomal RPGR promoter region upstream of a luciferase reporter gene dominant, autosomal recessive, X-linked, or mitochondrial were generated and analyzed by transient transfection and manner, but oligogenic inheritance has been established in a luciferase assays. Transgenic mouse lines carrying a 3-kb small proportion of families.3 X-linked RP (XLRP) is one of the human RPGR promoter sequence fused to lacZ were generated most consistently severe forms of RP, with a reported average and RPGR proximal promoter activity was analyzed by X-gal age at onset of 7.2 6 1.7 years.4 XLRP affects 10% to 20% of all staining. -

And Cone-Rod Dystrophy in Dogs
University of Pennsylvania ScholarlyCommons Departmental Papers (Vet) School of Veterinary Medicine 2012 RPGRIP1 and Cone-Rod Dystrophy in Dogs Tatyana N. Kuznetsova Barbara Zangerl University of Pennsylvania, [email protected] Gustavo D. Aguirre University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/vet_papers Part of the Eye Diseases Commons, Geriatrics Commons, Medical Biotechnology Commons, Medical Genetics Commons, Medical Immunology Commons, Ophthalmology Commons, and the Veterinary Medicine Commons Recommended Citation Kuznetsova, T. N., Zangerl, B., & Aguirre, G. D. (2012). RPGRIP1 and Cone-Rod Dystrophy in Dogs. Retinal Degenerative Diseases: Advances in Experimental Medicine and Biology, 723 321-328. http://dx.doi.org/ 10.1007/978-1-4614-0631-0_42 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/vet_papers/85 For more information, please contact [email protected]. RPGRIP1 and Cone-Rod Dystrophy in Dogs Abstract Cone–rod dystrophies (crd) represent a group of progressive inherited blinding diseases characterized by primary dysfunction and loss of cone photoreceptors accompanying or preceding rod death. Recessive crd type 1 was described in dogs associated with an RPGRIP1 exon 2 mutation, but with lack of complete concordance between genotype and phenotype. This review highlights role of the RPGRIP1, a component of complex protein networks, and its function in the primary cilium, and discusses the potential mechanisms of genotype–phenotype discordance observed in dogs with the RPGRIP1 mutation. Keywords RPGRIP1, polymorphism, cone-rod dystrophy, protein network, photoreceptor cilia Disciplines Eye Diseases | Geriatrics | Medical Biotechnology | Medical Genetics | Medical Immunology | Ophthalmology | Veterinary Medicine This journal article is available at ScholarlyCommons: https://repository.upenn.edu/vet_papers/85 Published in final edited form as: Adv Exp Med Biol. -

Selective Loss of RPGRIP1-Dependent Ciliary Targeting of NPHP4, RPGR and SDCCAG8 Underlies the Degeneration of Photoreceptor Neurons
Citation: Cell Death and Disease (2012) 3, e355; doi:10.1038/cddis.2012.96 & 2012 Macmillan Publishers Limited All rights reserved 2041-4889/12 www.nature.com/cddis Selective loss of RPGRIP1-dependent ciliary targeting of NPHP4, RPGR and SDCCAG8 underlies the degeneration of photoreceptor neurons H Patil1,3, N Tserentsoodol1,3, A Saha1, Y Hao1, M Webb1 and PA Ferreira*,1,2 The retinitis pigmentosa GTPase regulator (RPGR) and nephrocystin-4 (NPHP4) comprise two key partners of the assembly complex of the RPGR-interacting protein 1 (RPGRIP1). Mutations in RPGR and NPHP4 are linked to severe multisystemic diseases with strong retinal involvement of photoreceptor neurons, whereas those in RPGRIP1 cause the fulminant photoreceptor dystrophy, Leber congenital amaurosis (LCA). Further, mutations in Rpgrip1 and Nphp4 suppress the elaboration of the outer segment compartment of photoreceptor neurons by elusive mechanisms, the understanding of which has critical implications in uncovering the pathogenesis of syndromic retinal dystrophies. Here we show RPGRIP1 localizes to the photoreceptor connecting cilium (CC) distally to the centriole/basal body marker, centrin-2 and the ciliary marker, acetylated-a- tubulin. NPHP4 abuts proximally RPGRIP1, RPGR and the serologically defined colon cancer antigen-8 (SDCCAG8), a protein thought to partake in the RPGRIP1 interactome and implicated also in retinal–renal ciliopathies. Ultrastructurally, RPGRIP1 localizes exclusively throughout the photoreceptor CC and Rpgrip1nmf247 photoreceptors present shorter cilia with a ruffled membrane. Strikingly, Rpgrip1nmf247 mice without RPGRIP1 expression lack NPHP4 and RPGR in photoreceptor cilia, whereas the SDCCAG8 and acetylated-a-tubulin ciliary localizations are strongly decreased, even though the NPHP4 and SDCCAG8 expression levels are unaffected and those of acetylated-a-tubulin and c-tubulin are upregulated. -

An RPGRIP1 Mutation
Hindawi Publishing Corporation Stem Cells International Volume 2012, Article ID 685901, 12 pages doi:10.1155/2012/685901 Research Article Assessment of Hereditary Retinal Degeneration in the English Springer Spaniel Dog and Disease Relationship to an RPGRIP1 Mutation Kristina Narfstrom,¨ 1, 2 Manbok Jeong,3 Jennifer Hyman,4 Richard W. Madsen,5 and Tomas F. Bergstrom¨ 6 1 Department of Veterinary Medicine and Surgery, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA 2 Department of Ophthalmology, Mason Eye Institute, University of Missouri, Columbia, MO 65211, USA 3 Department of Veterinary Medicine and Surgery, College of Veterinary Medicine and BK21 Program for Veterinary Science, Seoul National University, Seoul 151-742, Republic of Korea 4 Eye Care For Animals, VA, 20176-3367, USA 5 Biostatistics, School of Medicine, University of Missouri, Columbia, MO 65211, USA 6 Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, 750 07 Uppsala, Sweden Correspondence should be addressed to Kristina Narfstrom,¨ [email protected] Received 12 September 2011; Revised 9 November 2011; Accepted 15 November 2011 Academic Editor: Henry J. Klassen Copyright © 2012 Kristina Narfstrom¨ et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Intensive breeding and selection on desired traits have produced high rates of inherited diseases in dogs. Hereditary retinal degeneration, often called progressive retinal atrophy (PRA), is prevalent in dogs with disease entities comparable to human retinitis pigmentosa (RP) and Leber’s congenital amaurosis (LCA). Recent molecular studies in the English Springer Spaniel (ESS) dog have shown that PRA cases are often homozygous for a mutation in the RPGRIP1 gene, the defect also causing human RP, LCA, and cone rod dystrophies. -

In Silico Approach to Calculate the Transcript Capacity
In silico approach to calculate the transcript capacity Young-Sup Lee, Kyung-Hye Won, Jae-Don Oh*, Donghyun Shin** Department of Animal Biotechnology, Chonbuk National University, Jeonju 54896, Korea Original article We sought the novel concept, transcript capacity (TC) and analyzed TC. Our approach to estimate TC was through an in silico method. TC refers to the capacity that a transcript ex- eISSN 2234-0742 erts in a cell as enzyme or protein function after translation. We used the genome-wide Genomics Inform 2019;17(3):e31 association study (GWAS) beta effect and transcription level in RNA-sequencing to esti- https://doi.org/10.5808/GI.2019.17.3.e31 mate TC. The trait was body fat percent and the transcript reads were obtained from the human protein atlas. The assumption was that the GWAS beta effect is the gene’s effect and TC was related to the corresponding gene effect and transcript reads. Further, we sur- Received: September 2, 2019 veyed gene ontology (GO) in the highest TC and the lowest TC genes. The most frequent Revised: September 18, 2019 GOs with the highest TC were neuronal-related and cell projection organization related. Accepted: September 19, 2019 The most frequent GOs with the lowest TC were wound-healing related and embryo devel- opment related. We expect that our analysis contributes to estimating TC in the diverse *Corresponding author: E-mail: [email protected] species and playing a benevolent role to the new bioinformatic analysis. **Corresponding author: Keywords: fat, genome-wide association study, in silico method, transcript capacity, RNA- E-mail: [email protected] seq Introduction There have been various experimental studies regarding enzyme activity [1,2]. -

Contribution of Structural and Intronic Mutations to RPGRIP1
bioRxiv preprint doi: https://doi.org/10.1101/211292; this version posted October 30, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Contribution of structural and intronic mutations to RPGRIP1- mediated inherited retinal dystrophies. Farzad Jamshidi1, Emily M. Place1, Daniel Navarro-Gomez1, Mathew Maher1, Elise Valkanas3, Monkol Lek2,3, Daniel MacArthur2,3, Kinga M. Bujakowska1, Eric A. Pierce1 1Ocular Genomics Institute, Department of Ophthalmology, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, MA. 2Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, Massachusetts, USA. 3Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA. Correspondence: Eric A. Pierce M.D., Ph.D. Solman and Libe Friedman Professor of Ophthalmology Massachusetts Eye and Ear Infirmary Harvard Medical School 243 Charles Street Boston, MA 02114 Phone: 617-573-6917 [email protected] bioRxiv preprint doi: https://doi.org/10.1101/211292; this version posted October 30, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1. Abstract With the completion of the first phase 3 human gene therapy randomized clinical trial, in the form of voretigene neparvovec for RPE65-mediated inherited retinal dystrophy, as well as the advent of more than 10 other gene therapy trials for inherited retinal disorders, accurate genetic diagnostics will have an increasingly important role in clinical decision- making. Current genetic diagnostic testing panels primarily focus on coding sequences. -

Photoreceptor Cilia and Retinal Ciliopathies
Downloaded from http://cshperspectives.cshlp.org/ on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Photoreceptor Cilia and Retinal Ciliopathies Kinga M. Bujakowska, Qin Liu, and Eric A. Pierce Ocular Genomics Institute, Massachusetts Eye and Ear Infirmary, Department of Ophthalmology, Harvard Medical School, Boston, Massachusetts 02114 Correspondence: [email protected] Photoreceptors are sensory neurons designed to convert light stimuli into neurological re- sponses. This process, called phototransduction, takes place in the outer segments (OS) of rod and cone photoreceptors. OS are specialized sensory cilia, with analogous structures to those present in other nonmotile cilia. Deficient morphogenesis and/or dysfunction of pho- toreceptor sensory cilia (PSC) caused by mutations in a variety of photoreceptor-specific and common cilia genes can lead to inherited retinal degenerations (IRDs). IRDs can manifest as isolated retinal diseases or syndromic diseases. In this review, we describe the structure and composition of PSC and different forms of ciliopathies with retinal involvement. We review the genetics of the IRDs, which are monogenic disorders but genetically diverse with regard to causality. hotoreceptors are sensory neurons designed morphogenesis and/or dysfunction of photore- Pto convert light stimuli into electrical re- ceptor sensory cilia (PSC) caused by mutations sponses, a process called phototransduction. in a variety of photoreceptor-specific and com- Phototransduction takes place in the highly spe- mon cilia genes can lead to a group of clinical cialized compartment of photoreceptors, the manifestations, called inherited retinal degener- outer segment (OS) (Pearring et al. 2013; Mol- ations (IRDs). In this review, we will discuss the day and Moritz 2015). -

Ciliary Genes in Renal Cystic Diseases
cells Review Ciliary Genes in Renal Cystic Diseases Anna Adamiok-Ostrowska * and Agnieszka Piekiełko-Witkowska * Department of Biochemistry and Molecular Biology, Centre of Postgraduate Medical Education, 01-813 Warsaw, Poland * Correspondence: [email protected] (A.A.-O.); [email protected] (A.P.-W.); Tel.: +48-22-569-3810 (A.P.-W.) Received: 3 March 2020; Accepted: 5 April 2020; Published: 8 April 2020 Abstract: Cilia are microtubule-based organelles, protruding from the apical cell surface and anchoring to the cytoskeleton. Primary (nonmotile) cilia of the kidney act as mechanosensors of nephron cells, responding to fluid movements by triggering signal transduction. The impaired functioning of primary cilia leads to formation of cysts which in turn contribute to development of diverse renal diseases, including kidney ciliopathies and renal cancer. Here, we review current knowledge on the role of ciliary genes in kidney ciliopathies and renal cell carcinoma (RCC). Special focus is given on the impact of mutations and altered expression of ciliary genes (e.g., encoding polycystins, nephrocystins, Bardet-Biedl syndrome (BBS) proteins, ALS1, Oral-facial-digital syndrome 1 (OFD1) and others) in polycystic kidney disease and nephronophthisis, as well as rare genetic disorders, including syndromes of Joubert, Meckel-Gruber, Bardet-Biedl, Senior-Loken, Alström, Orofaciodigital syndrome type I and cranioectodermal dysplasia. We also show that RCC and classic kidney ciliopathies share commonly disturbed genes affecting cilia function, including VHL (von Hippel-Lindau tumor suppressor), PKD1 (polycystin 1, transient receptor potential channel interacting) and PKD2 (polycystin 2, transient receptor potential cation channel). Finally, we discuss the significance of ciliary genes as diagnostic and prognostic markers, as well as therapeutic targets in ciliopathies and cancer. -
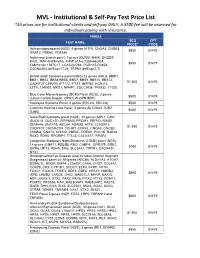
MVL - Institutional & Self-Pay Test Price List *All Prices Are for Institutional Clients and Self-Pay ONLY
MVL - Institutional & Self-Pay Test Price List *All prices are for institutional clients and self-pay ONLY. A $500 fee will be assessed for individuals paying with insurance. PANELS SEQ CPT TEST NAME PRICE* CODE Achromatopsiapanel (NGS), 6 genes (ATF6, CNGA3, CNGB3, $950 81479 GNAT2, PDE6C, PDE6H) Ashkenazi Jewish panel, 7 genes (CLRN1-N48K, DHDDS- K42E, MAK-K429insAlu, FAM161A-c.1355-6delCA, $550 81407 FAM161Ac.1567C>T, LCA5-Q279X, PCDH15-R245X, CACNA2D4-delExon17-26, TRPM1-delExon2-7) Bardet-Biedl Syndrome panel (NGS) 23 genes (ARL6, BBIP1, BBS1, BBS2, BBS4,BBS5, BBS7, BBS9, BBS10, BBS12, $1,500 81479 C8ORF37,CEP290, IFT172, IFT27, INPP5E, KCNJ13, LZTFL1,MKKS, MKS1, NPHP1, SDCCAG8, TRIM32, TTC8) Blue Cone Monochromacy (BCM) Panel (NGS), 2 genes $500 81479 (Locus Control Region, OPN1LW-OPN1MW) Cockayne Sydrome Panel, 2 genes (ERCC6, ERCC8) $500 81479 Common Hearing Loss Panel, 3 genes (SLC26A4, GJB2, $650 81479 GJB6) Cone-Rod Dystrophy panel (NGS), 33 genes (AIPL1, CRX, GUCA1A, GUCY2D, PITPNM3, PROM1, PRPH2, RIMS1, SEMA4A, UNC119, ABCA4, ADAM9, ATF6, C21ORF2, $1,950 81479 C8ORF37,CACNA2D4, CDHR1, CERKL, CNGA3, CNGB3, CNNM4, GNAT2, KCNV2, PDE6C, PDE6H, POC1B, RAB28, RAX2, RDH5, RPGRIP1, TTLL5, CACNA1F, RPGR) Congenital Stationary Night Blindness (CSNB) panel (NGS), 14 genes (GNAT1, PDE6B, RHO, CABP4, GPR179, GRK1, $950 81479 GRM6,LRIT3, RDH5, SAG, SLC24A1, TRPM1, CACNA1F, NYX) Developmental Eye Disease (also includes Anterior Segment Dysgenesis) panel v4: 59 genes (ABCB6, ALDH1A3, ATOH7, B3GALTL, BCOR, BMP4, c12orf57, CASK, CHD7, COL4A1, -

EGL Test Description
2460 Mountain Industrial Boulevard | Tucker, Georgia 30084 Phone: 470-378-2200 or 855-831-7447 | Fax: 470-378-2250 eglgenetics.com Ciliopathies: Deletion/Duplication Panel Test Code: DCIL1 Turnaround time: 2 weeks CPT Codes: 81406 x1, 81403 x1, 81405 x1 Condition Description The ciliopathies are a group of disorders caused by mutations in genes that encode proteins involved in the formation and function of cilia. Cilia are microtubule-based, hair-like cytoplasmic extensions that extend from the cell surface. The cilium is a highly conserved organelle that is structurally complex with approximately 1000 different recognized polypeptides. Cilia can be classified as either motile cilia or primary cilia (often called sensory cilia). Motile cilia, sometimes referred to as flagella, are typically found on epithelia cells that line the brain ventricles, oviducts, and respiratory tract. They can appear in bundles of 200-300 and can create movement of the extracellular fluid. Primary cilia are found on the surface of almost all cell types. They sense a wide variety of extracellular signals and transmit them to the interior of the cell. They are critical for developmental and physiological functions. Recent research suggests that motile cilia can be chemosensory as well. Cilia are a component of almost all cells, so defects in the cilia can lead to conditions that have features involving multiple organ systems, such as renal disease, cerebral anomalies, and retinal degeneration. Additional features include diabetes, skeletal dysplasia, obesity, and congenital fibrocystic diseases of the pancreas and liver; however, the specific phenotype depends on the specific cilia involved. Diseases tested by the panel include primary ciliary dyskinesia, nephronophthisis, Senior-Loken syndrome, Leber congenital amaurosis, Meckel- Gruber syndrome, Joubert and related syndromes, Bardet-Biedl syndrome, and many others. -

Disruption of Intraflagellar Protein Transport in Photoreceptor Cilia Causes Leber Congenital Amaurosis in Humans and Mice
Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice Karsten Boldt, … , Ronald Roepman, Marius Ueffing J Clin Invest. 2011;121(6):2169-2180. https://doi.org/10.1172/JCI45627. Research Article The mutations that cause Leber congenital amaurosis (LCA) lead to photoreceptor cell death at an early age, causing childhood blindness. To unravel the molecular basis of LCA, we analyzed how mutations in LCA5 affect the connectivity of the encoded protein lebercilin at the interactome level. In photoreceptors, lebercilin is uniquely localized at the cilium that bridges the inner and outer segments. Using a generally applicable affinity proteomics approach, we showed that lebercilin specifically interacted with the intraflagellar transport (IFT) machinery in HEK293T cells. This interaction disappeared when 2 human LCA-associated lebercilin mutations were introduced, implicating a specific disruption of IFT- dependent protein transport, an evolutionarily conserved basic mechanism found in all cilia. Lca5 inactivation in mice led to partial displacement of opsins and light-induced translocation of arrestin from photoreceptor outer segments. This was consistent with a defect in IFT at the connecting cilium, leading to failure of proper outer segment formation and subsequent photoreceptor degeneration. These data suggest that lebercilin functions as an integral element of selective protein transport through photoreceptor cilia and provide a molecular demonstration that disrupted IFT can lead to LCA. Find the latest version: https://jci.me/45627/pdf Related Commentary, page 2145 Research article Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice Karsten Boldt,1,2 Dorus A. -

Retinitis Pigmentosa Gtpase Regulator (RPGR) Protein Isoforms in Mammalian Retina: Insights Into X-Linked Retinitis Pigmentosa and Associated Ciliopathies
Vision Research 48 (2008) 366–376 www.elsevier.com/locate/visres Retinitis Pigmentosa GTPase Regulator (RPGR) protein isoforms in mammalian retina: Insights into X-linked Retinitis Pigmentosa and associated ciliopathies Shirley He a, Sunil K. Parapuram a, Toby W. Hurd b, Babak Behnam a,1, Ben Margolis b, Anand Swaroop a,c, Hemant Khanna a,* a Department of Ophthalmology and Visual Sciences, University of Michigan, W. K. Kellogg Eye Center, 1000 Wall Street, Ann Arbor, MI 48105, USA b Department of Internal Medicine, University of Michigan, Ann Arbor, MI 48105, USA c Department of Human Genetics, University of Michigan, Ann Arbor, MI 48105, USA Received 3 July 2007; received in revised form 3 August 2007 Abstract Mutations in the cilia-centrosomal protein Retinitis Pigmentosa GTPase Regulator (RPGR) are a frequent cause of retinal degener- ation. The RPGR gene undergoes complex alternative splicing and encodes multiple protein isoforms. To elucidate the function of major RPGR isoforms (RPGR1–19 and RPGRORF15), we have generated isoform-specific antibodies and examined their expression and local- ization in the retina. Using sucrose-gradient centrifugation, immunofluorescence and co-immunoprecipitation methods, we show that RPGR isoforms localize to distinct sub-cellular compartments in mammalian photoreceptors and associate with a number of cilia-cen- trosomal proteins. The RCC1-like domain of RPGR, which is present in all major RPGR isoforms, is sufficient to target it to the cilia and centrosomes in cultured cells. Our findings indicate that multiple isotypes of RPGR may perform overlapping yet somewhat distinct transport-related functions in photoreceptors. Ó 2007 Elsevier Ltd. All rights reserved.