Copyright by Sertan Kutal Gokce 2016
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UPC Platform Publisher Title Price Available 730865001347
UPC Platform Publisher Title Price Available 730865001347 PlayStation 3 Atlus 3D Dot Game Heroes PS3 $16.00 52 722674110402 PlayStation 3 Namco Bandai Ace Combat: Assault Horizon PS3 $21.00 2 Other 853490002678 PlayStation 3 Air Conflicts: Secret Wars PS3 $14.00 37 Publishers 014633098587 PlayStation 3 Electronic Arts Alice: Madness Returns PS3 $16.50 60 Aliens Colonial Marines 010086690682 PlayStation 3 Sega $47.50 100+ (Portuguese) PS3 Aliens Colonial Marines (Spanish) 010086690675 PlayStation 3 Sega $47.50 100+ PS3 Aliens Colonial Marines Collector's 010086690637 PlayStation 3 Sega $76.00 9 Edition PS3 010086690170 PlayStation 3 Sega Aliens Colonial Marines PS3 $50.00 92 010086690194 PlayStation 3 Sega Alpha Protocol PS3 $14.00 14 047875843479 PlayStation 3 Activision Amazing Spider-Man PS3 $39.00 100+ 010086690545 PlayStation 3 Sega Anarchy Reigns PS3 $24.00 100+ 722674110525 PlayStation 3 Namco Bandai Armored Core V PS3 $23.00 100+ 014633157147 PlayStation 3 Electronic Arts Army of Two: The 40th Day PS3 $16.00 61 008888345343 PlayStation 3 Ubisoft Assassin's Creed II PS3 $15.00 100+ Assassin's Creed III Limited Edition 008888397717 PlayStation 3 Ubisoft $116.00 4 PS3 008888347231 PlayStation 3 Ubisoft Assassin's Creed III PS3 $47.50 100+ 008888343394 PlayStation 3 Ubisoft Assassin's Creed PS3 $14.00 100+ 008888346258 PlayStation 3 Ubisoft Assassin's Creed: Brotherhood PS3 $16.00 100+ 008888356844 PlayStation 3 Ubisoft Assassin's Creed: Revelations PS3 $22.50 100+ 013388340446 PlayStation 3 Capcom Asura's Wrath PS3 $16.00 55 008888345435 -
Payday 2 64 Bit Patch
Payday 2 64 bit patch click here to download Jan 3, PAYDAY 2 Is there a 64 bit version exe of this game? Ok thanks, I found a way to patch it and make it run the 32 bit executable with 4Gb of. Apr 27, Hello! i can't run Payday 2 because i'm in 64 bit and i wan't to know how i I play this game on a 64bit os and I can confirm it only uses 4 gb of. Payday 2 64 Bit. Is there a 64 bit version exe of this game? Ok thanks, I found a way to patch it and make it run the 32 bit executable with 4Gb of Ram - and. May 3, Not really, no. It might become relevant if Payday 2 would for some reason start using more than the bit address space, but I don't think that's. Oct 12, Patch Method-. Why is it that when If it was 64 bit, then it would be quicker. Then once unpacked, its moved to the PAYDAY 2 directory. #7. Mar 3, Although this game sounds fun, Payday 2 has certain issues on Windows In order to fix Payday 2 crashes on Windows 10, it's advised that you run the locate the following line (it might look a bit different on your computer). Sep 13, Sad that my Tech is a bit nerfed, but that's probably because it was one of the best non-shotty weapons in the game. I think the SMG's still. May 17, This has the obvious advantage of only having to transmit tiny patch files. -

2021 Catalogue What Is Pinfinity?
2021 CATALOGUE WHAT IS PINFINITY? Pinfinity makes beautifully-crafted, collectible hard enamel pins, powered by their patented augmented reality platform. Fans can experience and unlock amazingly awesome content like, exclusive wallpapers, video, music, animations, selfie-frames, downloadables and more through the free Pinfinity app on Android and iOS. With more licenses, partnerships and exclusive promotions launching in 2022, there is something for every fan. Contents MAGIC: THE GATHERING pg 4-5 DUNGEONS & DRAGONS PG 6-7 STREET FIGHTER PG 8-9 POWER RANGERS PG 10-11 MY LITTLE PONY PG 12-13 WORMS PG 14-15 CRYPT OF THE NECRO DANCER PG 16 TETRIS PG 17 PAC-MAN PG 17 NASA PG 18 FUZZBALLS PG 19 LIMITED EDITION SETS 20-25 MAGIC: THE GATHERING PMTG001 Chandra, Pyromaster AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC PMTG002 Red Mana Crest AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC PMTG003 Jace, Memory Adept AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC PMTG004 Blue Mana Crest AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC PMTG005 Lilana of the Dark Realms AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC PMTG006 Black Mana Crest AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC PMTG007 Ajani, Caller of the Pride AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC PMTG008 White Mana Crest AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC PMTG009 Garruk, Caller of Beasts AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC PMTG010 Green Mana Crest AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC 30+ SKUS PLANNED FOR 2022 05 DUNGEONS & DRAGONS PDD001 Dragon Ampersand AR Pin MSRP: 12.99 Features: Artwork, Music, Selfie, DLC PDD002 Dungeon Master AR Pin MSRP: 12.99 Features: Animation, Music, Selfie, DLC. -
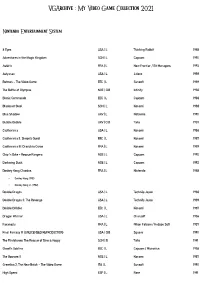
Vgarchive : My Video Game Collection 2021
VGArchive : My Video Game Collection 2021 Nintendo Entertainment System 8 Eyes USA | L Thinking Rabbit 1988 Adventures in the Magic Kingdom SCN | L Capcom 1990 Astérix FRA | L New Frontier / Bit Managers 1993 Astyanax USA | L Jaleco 1989 Batman – The Video Game EEC | L Sunsoft 1989 The Battle of Olympus NOE | CiB Infinity 1988 Bionic Commando EEC | L Capcom 1988 Blades of Steel SCN | L Konami 1988 Blue Shadow UKV | L Natsume 1990 Bubble Bobble UKV | CiB Taito 1987 Castlevania USA | L Konami 1986 Castlevania II: Simon's Quest EEC | L Konami 1987 Castlevania III: Dracula's Curse FRA | L Konami 1989 Chip 'n Dale – Rescue Rangers NOE | L Capcom 1990 Darkwing Duck NOE | L Capcom 1992 Donkey Kong Classics FRA | L Nintendo 1988 • Donkey Kong (1981) • Donkey Kong Jr. (1982) Double Dragon USA | L Technōs Japan 1988 Double Dragon II: The Revenge USA | L Technōs Japan 1989 Double Dribble EEC | L Konami 1987 Dragon Warrior USA | L Chunsoft 1986 Faxanadu FRA | L Nihon Falcom / Hudson Soft 1987 Final Fantasy III (UNLICENSED REPRODUCTION) USA | CiB Square 1990 The Flintstones: The Rescue of Dino & Hoppy SCN | B Taito 1991 Ghost'n Goblins EEC | L Capcom / Micronics 1986 The Goonies II NOE | L Konami 1987 Gremlins 2: The New Batch – The Video Game ITA | L Sunsoft 1990 High Speed ESP | L Rare 1991 IronSword – Wizards & Warriors II USA | L Zippo Games 1989 Ivan ”Ironman” Stewart's Super Off Road EEC | L Leland / Rare 1990 Journey to Silius EEC | L Sunsoft / Tokai Engineering 1990 Kings of the Beach USA | L EA / Konami 1990 Kirby's Adventure USA | L HAL Laboratory 1993 The Legend of Zelda FRA | L Nintendo 1986 Little Nemo – The Dream Master SCN | L Capcom 1990 Mike Tyson's Punch-Out!! EEC | L Nintendo 1987 Mission: Impossible USA | L Konami 1990 Monster in My Pocket NOE | L Team Murata Keikaku 1992 Ninja Gaiden II: The Dark Sword of Chaos USA | L Tecmo 1990 Rescue: The Embassy Mission EEC | L Infogrames Europe / Kemco 1989 Rygar EEC | L Tecmo 1987 Shadow Warriors FRA | L Tecmo 1988 The Simpsons: Bart vs. -

Worms Armageddon V3721 No Cd Crack
1 / 2 Worms Armageddon V3.7.2.1 No Cd Crack Generator Crack DayZ Keygen Steam FIFA 14 Crack Football . ... Il-2 sturmovik cliffs of dover trainer crysis 3 key generator simcityMua game pc b n quy n , mua ... Ativa a CD Key no cliente da Steam para fazer download do Rocket League. ... Tue, July 05, 2016 · worms armageddon v3.7.2.1 no-cd crack for.. Worms Armageddon V3.7.2.1 No-cd Crack For The Sims 2.. Worms Armageddon hasta el día de hoy es el mejor juego de Worms que haya ... No tenía idea que tuvieran cinemáticas los juegos D: Se ven lul xdxd ... .7.2.1_%5BWinXP-7-8%5D_%5BNoCD%5D_%5B2013%5D_%5BMulti%5D ... S: Instalás esto [url=ftp://ftp.team17.com/pub/t17/patches/pc/WAPatch.exe]Patch 3.0[/url] y .... Download worms armageddon patch for windows 7 J ai 2 versions différentes du jeu Worms Armageddon une installable et une 4) installer le patch 3.0, si il refuse ... US NO-CD (19.1KB) Search for related No-CD No-DVD Patch. ... Forza Motorsport Worms Armageddon v3.7.2.1 Patch Available NOW Get .... Download Worms armageddon serial crack, Request a new password if ... Worms: Armageddon v3.7.2.1 [EURO] No-CD/Fixed EXE- Worms: ... Worms Armageddon V3.7.2.1 No Cd Crack worms armageddon, worms armageddon controls, worms armageddon weapons, worms armageddon online, worms .... Make sure you are running the latest version (currently v3.7.2.1). Notice how they mention Windows 2000 support on the updates page linked below. -

Storm Staley
Storm Staley 2/15/01 Professor Lowood History of Videogame Design The Annelid Menace (The Worms Attack) “You had it coming!” This high-pitched admonishment manages to tell the computer game industry what it got hit by when the juggernaut that is Worms 2 burst onto the gaming scene in 1997. Worms 2 is a very simple but extremely addictive computer game that proves that fun doesn’t necessarily require the latest in graphics technology or complicated gameplay. In the process it manages to give you a totally new perspective on life, the universe and – well, no, actually, it just sucks you in and engages you for hours on end. Worms 2 was developed by Team17 Software, a small software company based in Ossett, England (the wacky British humor connection is thus made clear) and published in late 1997 by MicroProse Software. The creator of the Worms series is Andy Davidson, now a cult legend for his work on the series and his general nuttiness. An interview with Andy on the Team17 website reveals this unforgettable quote: “As a child I was convinced that the stone donkey in the garden was a real donkey that my parents had covered in concrete. I therefore spent quite a bit of time chipping away at its tail to see if there was fur under there. This probably explains the state I’m in today!”1 The game was produced by Martyn Brown, with artists Rico Holmes and Danny Cartwright (creating the rich landscapes and the animation of the worms in the game, which according to the website is composed of over 14,000 animations2), and main programmers Karl Morton, Phil Carlisle, and Colin Surridge, with Chris Blyth creating the game’s full motion video scenes. -

GOG-API Documentation Release 0.1
GOG-API Documentation Release 0.1 Gabriel Huber Jun 05, 2018 Contents 1 Contents 3 1.1 Authentication..............................................3 1.2 Account Management..........................................5 1.3 Listing.................................................. 21 1.4 Store................................................... 25 1.5 Reviews.................................................. 27 1.6 GOG Connect.............................................. 29 1.7 Galaxy APIs............................................... 30 1.8 Game ID List............................................... 45 2 Links 83 3 Contributors 85 HTTP Routing Table 87 i ii GOG-API Documentation, Release 0.1 Welcome to the unoffical documentation of the APIs used by the GOG website and Galaxy client. It’s a very young project, so don’t be surprised if something is missing. But now get ready for a wild ride into a world where GET and POST don’t mean anything and consistency is a lucky mistake. Contents 1 GOG-API Documentation, Release 0.1 2 Contents CHAPTER 1 Contents 1.1 Authentication 1.1.1 Introduction All GOG APIs support token authorization, similar to OAuth2. The web domains www.gog.com, embed.gog.com and some of the Galaxy domains support session cookies too. They both have to be obtained using the GOG login page, because a CAPTCHA may be required to complete the login process. 1.1.2 Auth-Flow 1. Use an embedded browser like WebKit, Gecko or CEF to send the user to https://auth.gog.com/auth. An add-on in your desktop browser should work as well. The exact details about the parameters of this request are described below. 2. Once the login process is completed, the user should be redirected to https://www.gog.com/on_login_success with a login “code” appended at the end. -

Microsoft Xbox Live Arcade
Microsoft Xbox Live Arcade Last Updated on September 27, 2021 Title Publisher Qty Box Man Comments 0 Day Attack on Earth Square Enix 0-D: Beat Drop Arc System Works 1942: Joint Strike Capcom 3 on 3 NHL Arcade EA Freestyle 3D Ultra Minigolf Adventures Sierra Online 3D Ultra Minigolf Adventures 2 Konami Abyss Odyssey Atlus Aces of the Galaxy Artech Studios Adventures of Shuggy, The Valcon Games Aegis Wing Microsoft After Burner Climax Sega Age of Booty Capcom AirMech Arena Ubisoft Alan Wake's American Nightmare Microsoft Alein Spidey Kalypso Media Alien Breed 2: Assault Team17 Alien Breed 3: Descent Team 17 Alien Breed Evolution: Episode 1 Team 17 Alien Hominid HD The Behemoth Alien Spidy Kalypso Media All Zombies Must Die! Square Enix Altered Beast Sega American Mensa Academy Square Enix Amy VectorCell Ancients of Ooga Microsoft Anomaly: Warzone Earth Microsoft Apples to Apples THQ Aqua Xbox LIVE Arcade Are You Smarter Than A 5th Grader? THQ Arkadian Warriors Sierra Online ARKANOID Live! Xbox LIVE Arcade Ascend: Hand of Kul Microsoft Studios Assassin's Creed: Liberation HD Ubisoft Assault Heroes Sierra Online Assault Heroes 2 Sierra Online Asteroids & Asteroids Deluxe Atari AstroPop Oberon Media Awesomenauts DTP Entertainment Axel & Pixel 2K Games Babel Rising Ubisoft Backbreaker Vengence 505 Games Band of Bugs NinjaBee Bang Bang Racing Digital Reality Software Bangai-O HD: Missile Fury D3 Publisher Banjo-Kazooie Microsoft Banjo-Tooie Microsoft Bankshot Billiards 2 PixelStorm Bastion Warner Bros. Interactive Batman: Arkham Origins Blackgate - Deluxe Edition Warner Bros. Interactive En... Battle: Los Angeles Konami BattleBlock Theater Microsoft Battlefield 1943 Electronic Arts Battlestar Galactica Sierra Online Battlezone Atari This checklist is generated using RF Generation's Database This checklist is updated daily, and it's completeness is dependent on the completeness of the database. -
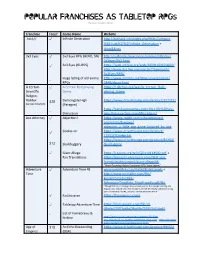
POPULAR FRANCHISES AS TABLETOP RPGS Revision 10 (Apr.2021)
POPULAR FRANCHISES AS TABLETOP RPGS Revision 10 (Apr.2021) Franchise Free? Game Name Website .hack// ✓ Infinite Generation http://dothack.info/index.php?title=Category %3A.hack%2F%2FInfinite_Generation + Odds&Ends 3x3 Eyes ✓ 3x3 Eyes RPG (HERO, SN) http://surbrook.devermore.com/worldbooks/ 3x3eyes/3x3.html ✓ 3x3 Eyes (GURPS) https://web.archive.org/web/20001109020600/ http://www.dca.fee.unicamp.br/~henriqmh/ 3x3Eyes/RPG/ Huge listing of old anime http://www.oocities.org/timessquare/arena/ - RPGs 2448/about.html A Certain ✓ A Certain Roleplaying https://1d4chan.org/wiki/A_Certain_Role- Scientific Game playing_Game Railgun, Raildex $20 Demongate High https://www.drivethrurpg.com/product/127312/ See also Smallville. (Paragon) https://yarukizerogames.com/2011/02/10/role- - Discussion play-this-a-certain-scientific-railgun/ Ace Attorney ✓ Abjection! https://www.reddit.com/r/AceAttorney/ comments/8zwgwa/ abjection_a_little_rpg_game_inspired_by_ace ✓ Cookie Jar https://www.drivethrurpg.com/product/ 115517/Cookie-Jar https://www.drivethrurpg.com/product/84260/ $12 Skullduggery Skulduggery ✓ Giant Allege: https://i.4pcdn.org/tg/1520114144592.pdf + Fan Translations https://projects.inklesspen.com/fatal-and- friends/professorprof/giant-allege/#6 “Heart-Pounding Robot Courtroom RPG" from Japan!” Adventure ✓ Adventure Time 4E www.mediafire.com/?ia9628cdx6zacwb + Time http://www.mediafire.com/file/ kmxkml5mzduc994/ AdventureTimeBeta_PrintFriendly.pdf/file “Though I’m not releasing a new version just for this, people reading this original post should -

Worms Armageddon Crack Unlock Code
1 / 4 Worms Armageddon Crack Unlock Code Worms 2 Armageddon v1.4.1 APK Free Download latest version for android. So free download full APK of Worms 2: Armageddon v1.4.0 for ... This is single direct link of Worms 2 Armageddon v1.4.0 APK Mod Unlocked With .... Get all the inside info, cheats, hacks, codes, walkthroughs for Worms Armageddon on GameSpot. ... allowing you to earn a gold medal in that mission and unlocking a new weapon. ... In-Depth Guides, Wormnet/Patch Guide by Edge AoM, 22K .... Patch do gry Worms Armageddon poniżej anglojęzyczny opis ... include a crack, serial number, unlock code, cd key or keygen key generator.. Worms Armageddon cheats, Passwords, and Codes for GBC. Jump to: Password (2). Show by ... Enter on the Landscape selection menu to unlock new worlds.. My friends, I am glad to present you a free Code Generator for Borderlands The Handsome Collection ... Borderlands GOTY Enhanced (+1 Trainer) [Cheat Happens] Unlock more options including ... Unlimited Golden Keys in BL:GOTY Enhanced on PC cheat/hack. ... Worms 2 Armageddon GOTY. bin back into the default.. ... 60th Anniversary, enjoy 50% off the in-game premium unlock from December 4 ... TORRENT download. downloads (7 days) 2 Sep 08, 2010 · Crazy Taxi 3Full PC ... 8 cheat codes and secrets, 1 review, 1 critic review, and 29 user screenshots. ... Worms Armageddon; Pilotii Nascar; Prince Of Persia; Download Blackwake; .... Crysis 2 Patch, DX11 and HD Texture Pack This patch fixes a number of gameplay issues and bugs ... Crysis 2 Code Generator 1.0 key code generator: Motorola Unlocking Code Generator .. -
Worms 1995 Download
Worms 1995 Download Worms 1995 Download 1 / 2 Most people looking for Worms 3D for windows 10 downloaded: Worms 3D ... Activate Windows 10 Worms has been knocking about since 1995, and with each .... Worms is an DOS strategy game, developed by Team 17, designed by Andy Davidson and published by Ocean Software in 1995. It's available for download.. Free family feud download for mac. If you are ... Worms 1. Released: Jan 1, 1995; Size: 1.72 MB. Downloads: 4,301; Rating: Tested: Free from .... Worms 2 Armageddon Free Download for PC is anartillery strategy game ... Worms (1995) • It was first released on Xbox Live Arcade for 800 Microsoft points on .... Worms Armageddon Download Free download Worms Armageddon (138 Mb) Get The Full ... Worms MS-DOS game released by Team 17 Software in 1995. Abandonware game Worms: Armageddon is a strategy game released in 1999 ... Why can't I download the game? worms armageddon is protected by copyright.. It is part of the Worms series and a sequel to the 1995 game Worms. ... Worms 2 Armageddon Games Latest Download For PC Windows Full .... Worms 0.0.95 apk free Download - ApkHere.com - Mobile. ... 35.58 MB. Price: [free]; Version: 0.0.95 Report a new version; Category: Casual .... Worms (1995) • Worms Open warfare 2 is a strategy game where you control a ... Worms World Party (Remastered) Download the Worms - Open Warfare (EU) ... worms worms, worms game, worms armageddon, worms in humans, worms wmd, worms online, worms world party, worms rumble, worms ultimate mayhem, worms reloaded, worms 4, worms battlegrounds, worms in children Worms is an artillery strategy video game developed by Team17 and released in 1995. -

June 29, 2015 by Electronic Mail Jacqueline C. Charlesworth
June 29, 2015 By Electronic Mail Jacqueline C. Charlesworth General Counsel and Associate Register of Copyrights U.S. Copyright Office Library of Congress 101 Independence Ave SE Washington, DC 20559-6000 Re: Docket No. 2014-7, Proposed Class 23: Abandoned Software–Video Games Requiring Server Communication Dear Ms. Charlesworth: On behalf of the Entertainment Software Association (“ESA”), this letter responds to your June 3, 2015 letter regarding Proposed Class 23. 1. Please explain whether, and under what circumstances, video game publishers reissue or repackage games where the publisher or developer has previously ended support for a server that enables single-player and/or multiplayer play. Please provide illustrative examples, including an explanation of the similarities and differences between the original and reissued products and the role of technological protection measures. How frequently does this occur? A video game publisher may invest millions of dollars developing a single video game. In order to obtain a return on this significant investment, publishers are continuously looking for ways to refresh these highly-valuable copyrighted works during their copyright terms, adapt them for different platforms, and reissue games after server support has ended. The following examples illustrate just a few of the ways in which publishers have reissued or repackaged their copyrighted video games:1 1 See also “E3 2015: Old Games Find New Life,” The Times of India (June 17, 2015), http://timesofindia.indiatimes.com/tech/gaming/E3-2015-Old-games-find-new- life/articleshow/47704590.cms. Jacqueline C. Charlesworth June 29, 2015 Page 2 • Worms was first released in 1995 as an artillery strategy game for personal computers with single-player and online multi-player functionality.