Germline Polymorphisms in RNF31 Regulate Linear Ubiquitination And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genome-Wide Sirna Screen for Mediators of NF-Κb Activation
Genome-wide siRNA screen for mediators SEE COMMENTARY of NF-κB activation Benjamin E. Gewurza, Fadi Towficb,c,1, Jessica C. Marb,d,1, Nicholas P. Shinnersa,1, Kaoru Takasakia, Bo Zhaoa, Ellen D. Cahir-McFarlanda, John Quackenbushe, Ramnik J. Xavierb,c, and Elliott Kieffa,2 aDepartment of Medicine and Microbiology and Molecular Genetics, Channing Laboratory, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115; bCenter for Computational and Integrative Biology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114; cProgram in Medical and Population Genetics, The Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02142; dDepartment of Biostatistics, Harvard School of Public Health, Boston, MA 02115; and eDepartment of Biostatistics and Computational Biology and Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA 02115 Contributed by Elliott Kieff, December 16, 2011 (sent for review October 2, 2011) Although canonical NFκB is frequently critical for cell proliferation, (RIPK1). TRADD engages TNFR-associated factor 2 (TRAF2), survival, or differentiation, NFκB hyperactivation can cause malig- which recruits the ubiquitin (Ub) E2 ligase UBC5 and the E3 nant, inflammatory, or autoimmune disorders. Despite intensive ligases cIAP1 and cIAP2. CIAP1/2 polyubiquitinate RIPK1 and study, mammalian NFκB pathway loss-of-function RNAi analyses TRAF2, which recruit and activate the K63-Ub binding proteins have been limited to specific protein classes. We therefore under- TAB1, TAB2, and TAB3, as well as their associated kinase took a human genome-wide siRNA screen for novel NFκB activa- MAP3K7 (TAK1). TAK1 in turn phosphorylates IKKβ activa- tion pathway components. Using an Epstein Barr virus latent tion loop serines to promote IKK activity (4). -

Whole Genome Sequencing of One Complex Pedigree Illustrates Challenges with Genomic Medicine Han Fang1,2,3†, Yiyang Wu1,2†, Hui Yang7,8, Margaret Yoon1, Laura T
Fang et al. BMC Medical Genomics (2017) 10:10 DOI 10.1186/s12920-017-0246-5 RESEARCH ARTICLE Open Access Whole genome sequencing of one complex pedigree illustrates challenges with genomic medicine Han Fang1,2,3†, Yiyang Wu1,2†, Hui Yang7,8, Margaret Yoon1, Laura T. Jiménez-Barrón1,4, David Mittelman5, Reid Robison5,6, Kai Wang7,9,10,11 and Gholson J. Lyon1,2,6* Abstract Background: Human Phenotype Ontology (HPO) has risen as a useful tool for precision medicine by providing a standardized vocabulary of phenotypic abnormalities to describe presentations of human pathologies; however, there have been relatively few reports combining whole genome sequencing (WGS) and HPO, especially in the context of structural variants. Methods: We illustrate an integrative analysis of WGS and HPO using an extended pedigree, which involves Prader–Willi Syndrome (PWS), hereditary hemochromatosis (HH), and dysautonomia-like symptoms. A comprehensive WGS pipeline was used to ensure reliable detection of genomic variants. Beyond variant filtering, we pursued phenotypic prioritization of candidate genes using Phenolyzer. Results: Regarding PWS, WGS confirmed a 5.5 Mb de novo deletion of the parental allele at 15q11.2 to 15q13.1. Phenolyzer successfully returned the diagnosis of PWS, and pinpointed clinically relevant genes in the deletion. Further, Phenolyzer revealed how each of the genes is linked with the phenotypes represented by HPO terms. For HH, WGS identified a known disease variant (p.C282Y) in HFE of an affected female. Analysis of HPO terms alone fails to provide a correct diagnosis, but Phenolyzer successfully revealed the phenotype-genotype relationship using a disease-centric approach. -

Diagnostic Interpretation of Genetic Studies in Patients with Primary
AAAAI Work Group Report Diagnostic interpretation of genetic studies in patients with primary immunodeficiency diseases: A working group report of the Primary Immunodeficiency Diseases Committee of the American Academy of Allergy, Asthma & Immunology Ivan K. Chinn, MD,a,b Alice Y. Chan, MD, PhD,c Karin Chen, MD,d Janet Chou, MD,e,f Morna J. Dorsey, MD, MMSc,c Joud Hajjar, MD, MS,a,b Artemio M. Jongco III, MPH, MD, PhD,g,h,i Michael D. Keller, MD,j Lisa J. Kobrynski, MD, MPH,k Attila Kumanovics, MD,l Monica G. Lawrence, MD,m Jennifer W. Leiding, MD,n,o,p Patricia L. Lugar, MD,q Jordan S. Orange, MD, PhD,r,s Kiran Patel, MD,k Craig D. Platt, MD, PhD,e,f Jennifer M. Puck, MD,c Nikita Raje, MD,t,u Neil Romberg, MD,v,w Maria A. Slack, MD,x,y Kathleen E. Sullivan, MD, PhD,v,w Teresa K. Tarrant, MD,z Troy R. Torgerson, MD, PhD,aa,bb and Jolan E. Walter, MD, PhDn,o,cc Houston, Tex; San Francisco, Calif; Salt Lake City, Utah; Boston, Mass; Great Neck and Rochester, NY; Washington, DC; Atlanta, Ga; Rochester, Minn; Charlottesville, Va; St Petersburg, Fla; Durham, NC; Kansas City, Mo; Philadelphia, Pa; and Seattle, Wash AAAAI Position Statements,Work Group Reports, and Systematic Reviews are not to be considered to reflect current AAAAI standards or policy after five years from the date of publication. The statement below is not to be construed as dictating an exclusive course of action nor is it intended to replace the medical judgment of healthcare professionals. -

Identifying Main Genes and Pathways by Using Gene Expression Profiling in Primary Immunodeficiency HOIL-1/RBCK1 Disorder Patients
ORIGINAL ARTICLE Identifying Main Genes and Pathways by Using Gene Expression Profiling in Primary Immunodeficiency HOIL-1/RBCK1 Disorder Patients Khyber Shinwari1, Guojun Liu2, Mikhail Bolkov3, Monib Ullah4, Irina Tuzankina3 1 Department of Immunochemistry, Institute of Chemical Engineering, Ural Federal University, Yekaterinburg, Russia 2 School of Life Science and Technology, Inner Mongolia University of Science and Technology, Baotou 014010, China 3 Institute of Immunology and Physiology of the Ural Branch of the Russian Academy of Sciences, Yekaterinburg, Russia 4 Institute of Microbiology, University of Agriculture Faisalabad, Faisalabad, Pakistan Received: 16 Sep. 2020; Accepted: 28 Mar. 2021 Abstract- HOIL-1/RBCK1 deficiency is a new autosomal receiving disorder with dysfunctional cellular responses to pro-inflammatory cytokines, leading to auto-inflammation, pyogenic bacterial disease, and muscle amylopectinosis growth. Our study with integrated bioinformatics studies of the feature genes and the correlative gene functions, investigated the molecular mechanisms of RBCK1 deficiency. GSE31064 dataset expression profile was downloaded from the Omnibus Gene Expression database. Between RBCK1, MYDK88, NEMO deficient fibroblast, and healthy fibroblast specimens, differentially expressed genes (DEGs) were defined. Gene ontology (GO) gene role enrichment analysis and the Kyoto Encyclopedia of Gene and Genome (KEGG) pathway analysis were performed using the Annotation, Visualization and Integrated Discovery Database (DAVID). The protein-protein -

Towards Personalized Medicine in Psychiatry: Focus on Suicide
TOWARDS PERSONALIZED MEDICINE IN PSYCHIATRY: FOCUS ON SUICIDE Daniel F. Levey Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the Program of Medical Neuroscience, Indiana University April 2017 ii Accepted by the Graduate Faculty, Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Andrew J. Saykin, Psy. D. - Chair ___________________________ Alan F. Breier, M.D. Doctoral Committee Gerry S. Oxford, Ph.D. December 13, 2016 Anantha Shekhar, M.D., Ph.D. Alexander B. Niculescu III, M.D., Ph.D. iii Dedication This work is dedicated to all those who suffer, whether their pain is physical or psychological. iv Acknowledgements The work I have done over the last several years would not have been possible without the contributions of many people. I first need to thank my terrific mentor and PI, Dr. Alexander Niculescu. He has continuously given me advice and opportunities over the years even as he has suffered through my many mistakes, and I greatly appreciate his patience. The incredible passion he brings to his work every single day has been inspirational. It has been an at times painful but often exhilarating 5 years. I need to thank Helen Le-Niculescu for being a wonderful colleague and mentor. I learned a lot about organization and presentation working alongside her, and her tireless work ethic was an excellent example for a new graduate student. I had the pleasure of working with a number of great people over the years. Mikias Ayalew showed me the ropes of the lab and began my understanding of the power of algorithms. -

The Role of Malt1 in T Cell Immunity
THE ROLE OF MALT1 IN T CELL IMMUNITY by Sohyeong Kang B.Sc., The University of Toronto, 2014 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (PATHOLOGY AND LABORATORY MEDICINE) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) October 2017 © Sohyeong Kang, 2017 Abstract Individuals with combined immunodeficiency exhibit near-normal frequencies of T and B cells but suffer from severe recurrent infections related to defective cellular or humoral immunity. A child admitted to BC Children’s Hospital displayed a novel clinical presentation of combined immunodeficiency with immune dysregulation. The patient was found to be homozygous for missense mutations in the MALT1 gene inherited from her consanguineous parents. MALT1 plays critical roles in activating the NF-κB pathway upon T cell and B cell receptor stimulation. MALT1 functions by at least two different mechanisms: (1) as a scaffolding molecule bringing CARD11 and BCL-10 to form the CBM signalosome complex and (2) as a caspase-like protease cleaving substrates to regulate NF-κB signaling. Studies in Malt1−/− mice have revealed weakened T cell immunity whereas mice expressing MALT1 lacking paracaspase activity (but normal scaffolding function) are prone to fetal autoimmunity. In addition, patients with MALT1 mutations shared similar clinical characteristics, experiencing chronic infections and gastrointestinal inflammation. Together, these findings raise questions regarding the nature of our patient’s mutant MALT1 protein and the role it plays in her pro-inflammatory phenotype. Our data have shown that MALT1 is essential for IL-2 production in CD4 T cells despite the dispensable role in regulatory T cell development. -

Linkage to 20P13 Including the ANGPT4 Gene in Families With
Journal of Human Genetics (2010) 55, 649–655 & 2010 The Japan Society of Human Genetics All rights reserved 1434-5161/10 $32.00 www.nature.com/jhg ORIGINAL ARTICLE Linkageto20p13includingtheANGPT4 gene in families with mixed Alzheimer’s disease and vascular dementia Anna Sille´n1, Jesper Brohede1, Lena Lilius1, Charlotte Forsell1, Jorge Andrade2,5, Jacob Odeberg2, Hayao Ebise3, Bengt Winblad1 and Caroline Graff1,4 This study aimed at identifying novel susceptibility genes for a mixed phenotype of Alzheimer’s disease and vascular dementia. Results from a genome scan showed strongest linkage to 20p13 in 18 families, and subsequent fine mapping was performed with both microsatellites and single-nucleotide polymorphisms in 18 selected candidate transcripts in an extended sample set of 30 families. The multipoint linkage peak was located at marker rs2144151 in the ANGPT4 gene, which is a strong candidate gene for vascular disease because of its involvement in angiogenesis. Although the significance of the linkage decreased, we find this result intriguing, considering that we included additional families, and thus the reduced linkage signal may be caused by genetic heterogeneity. Journal of Human Genetics (2010) 55, 649–655; doi:10.1038/jhg.2010.79; published online 1 July 2010 Keywords: Alzheimer’s disease; ANGPT4; association study; candidate genes; familial dementia; linkage analysis; SNP INTRODUCTION Previous genome-wide family-based linkage analyses have identified Alzheimer’s disease (AD; MIM104300) is the major cause of dementia several loci linked to AD9–19 and confirmed linkage by several in the elderly. Its main neuropathological signs are extracellular groups has been reported to chromosome (chr.) 9p, 9q, 10q, 12p depositions of amyloid b-protein and intracellular neurofibrillary and 19q.20–24 Extensive follow-up and fine mapping studies have been tangles composed of hyperphosphorylated tau. -

Linear Ubiquitination Is Involved in the Pathogenesis of Optineurin-Associated Amyotrophic Lateral Sclerosis
ARTICLE Received 10 Sep 2015 | Accepted 12 Jul 2016 | Published 24 Aug 2016 DOI: 10.1038/ncomms12547 OPEN Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis Seshiru Nakazawa1,2,*, Daisuke Oikawa1,3,*, Ryohei Ishii4,*, Takashi Ayaki5,6, Hirotaka Takahashi7, Hiroyuki Takeda7, Ryuichiro Ishitani4, Kiyoko Kamei1, Izumi Takeyoshi2, Hideshi Kawakami8, Kazuhiro Iwai9, Izuho Hatada10, Tatsuya Sawasaki7, Hidefumi Ito5, Osamu Nureki4 & Fuminori Tokunaga1,3 Optineurin (OPTN) mutations cause neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and glaucoma. Although the ALS-associated E478G mutation in the UBAN domain of OPTN reportedly abolishes its NF-kB suppressive activity, the precise molecular basis in ALS pathogenesis still remains unclear. Here we report that the OPTN-UBAN domain is crucial for NF-kB suppression. Our crystal structure analysis reveals that OPTN-UBAN binds linear ubiquitin with homology to NEMO. TNF-a-mediated NF-kB activation is enhanced in OPTN-knockout cells, through increased ubiquitination and association of TNF receptor (TNFR) complex I components. Furthermore, OPTN binds caspase 8, and OPTN deficiency accelerates TNF-a-induced apoptosis by enhancing complex II formation. Immunohistochemical analyses of motor neurons from OPTN-associated ALS patients reveal that linear ubiquitin and activated NF-kB are partially co-localized with cytoplasmic inclusions, and that activation of caspases is elevated. Taken together, OPTN regulates both NF-kB activation and apoptosis via linear ubiquitin binding, and the loss of this ability may lead to ALS. 1 Laboratory of Molecular Cell Biology, Institute for Molecular and Cellular Regulation, Gunma University, 3-39-15 Showa-machi, Maebashi, Gunma 371-8512, Japan. -
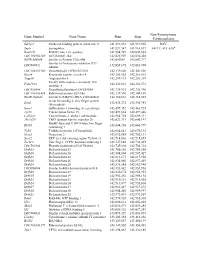
Gene Symbol Gene Name Start Stop Non-Synonymous Polymorphism
Non-Synonymous Gene Symbol Gene Name Start Stop Polymorphism Sdcbp2 Syndecan binding protein (syntenin) 2 141,891,232 141,919,905 I82V Snph Syntaphilin 141,921,547 141,961,697 A411T, 415_420* Rad21l1 RAD21-like 1 (S. pombe) 141,984,729 142,009,665 LOC100363280 mCG140681-like 142,022,959 142,030,458 RGD1564048 Similar to Protein C20orf46 142,043961 142,047,717 Similar to Proteasome inhibitor PI31 LOC689852 142,058,695 142,083,908 subunit LOC100363380 Hypothetical LOC100363380 142,139,020 142,143,826 Rspo4 R-spondin family, member 4 142,185,028 142,216,015 Angpt4 Angiopoietin 4 142,249,115 142,282,307 Family with sequence similarity 110, Fam110a 142,326,013 142,328,572 member A LOC689884 Hypothetical protein LOC689884 142,335,923 142,336,396 LOC100363434 Ribosomal protein S29-like 142,339,596 142,341,169 RGD1304644 Similar to RIKEN cDNA 2310046K01 142,360,661 142,365,285 Scratch homolog 2, zinc finger protein Scrt2 142,434,272 142,445,984 (Drosophila) Srxn1 Sulfiredoxin 1 homolog (S. cerevisiae) 142,457,352 142,462,725 Tcf15 Transcription factor 15 142,491,664 142,497,446 Csnk2a1 Casein kinase 2, alpha 1 polypeptide 142,564,754 142,609,311 Tbc1d20 TBC1 domain family, member 20 142,623,713 142,640,197 RanBP-type and C3HC4-type zinc finger Rbck1 142,644,156 142,660,799 containing 1 Trib3 Tribbles homolog 3 (Drosophila) 142,664,641 142,670,135 Nrsn2 Neurensin 2 142,692,988 142,702,131 Sox12 SRY (sex determining region Y)-box 12 142,714,836 142,715,855 Zcchc3 Zinc finger, CCHC domain containing 3 142,727,648 142,730,465 LOC502684 Hypothetical -

Uncovering Ubiquitylation Pathways in Liver Metabolism by a Proteomic Approach Helena Magliarelli
Uncovering ubiquitylation pathways in liver metabolism by a proteomic approach Helena Magliarelli To cite this version: Helena Magliarelli. Uncovering ubiquitylation pathways in liver metabolism by a proteomic approach. Molecular biology. Université de Strasbourg, 2014. English. NNT : 2014STRAJ083. tel-01413594 HAL Id: tel-01413594 https://tel.archives-ouvertes.fr/tel-01413594 Submitted on 10 Dec 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. UNIVERSITÉ DE STRASBOURG École Doctorale des Sciences de la Vie et de la Santé IGBMC - Institut de Génétique et de Biologie Moléculaire et Cellulaire THÈSE présentée par : Helena MAGLIARELLI soutenue le : 09 Octobre 2014 pour obtenir le grade de : !"#$%&'($')R%+,-$&.,# '($'0#&1.2!%&3 Discipline/ Spécialité : Aspects moléculaires et cellulaires de la biologie Uncovering ubiquitylation pathways in liver metabolism by a proteomic approach Mr RICCI Romeo Directeur de thèse Mr BAUMERT Thomas Examinateur Mme PICHLER Andrea Rapporteur externe Mme POSTIC Catherine Rapporteur externe Acknowledgements Acknowledgements First of all, I would like to thank Romeo for giving me always the very best opportunities to develop and become a better scientist. Thank you for guiding me throught the path that has brought me to this point. -

Neonatal-Onset Autoinflammation and Immunodeficiency Caused by Heterozygous Missense Mutation of the Proteasome Subunit Β-Type 9
medRxiv preprint doi: https://doi.org/10.1101/2021.02.01.21250077; this version posted February 2, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Neonatal-onset autoinflammation and immunodeficiency caused by heterozygous missense mutation of the proteasome subunit β-type 9 Nobuo Kanazawa1,2*, Hiroaki Hemmi3,4*, Noriko Kinjo5, Hidenori Ohnishi6, Jun Hamazaki7, Hiroyuki Mishima8, Akira Kinoshita8, Tsunehiro Mizushima9, Satoru Hamada5, Kazuya Hamada5, Norio Kawamoto6, Saori Kadowaki6, Yoshitaka Honda10, Kazushi Izawa10, Ryuta Nishikomori11, Miyuki Tsumura12, Yusuke Yamashita13, Shinobu Tamura13, Takashi Orimo3,14, Toshiya Ozasa3, Takashi Kato3, Izumi Sasaki3, Yuri Fukuda-Ohta3, Naoko Wakaki-Nishiyama3, Yutaka Inaba1, Kayo Kunimoto1, Satoshi Okada12, Takeshi Taketani15, Koichi Nakanishi5, Shigeo Murata7, Koh-ichiro Yoshiura8, Tsuneyasu Kaisho3 1 Department of Dermatology, Wakayama Medical University, Wakayama, Japan. 2Department of Dermatology, Hyogo College of Medicine, Hyogo, Japan. 3Department of Immunology, Institute of Advanced Medicine, Wakayama Medical University, Wakayama, Japan. 4Laboratory of Immunology, Faculty of Veterinary Medicine, Okayama University of Science, Ehime, Japan. 5Department of Child Health and Welfare (Pediatrics), Graduate School of Medicine, University of the Ryukyus, Okinawa, Japan 6Department of Pediatrics, Graduate School