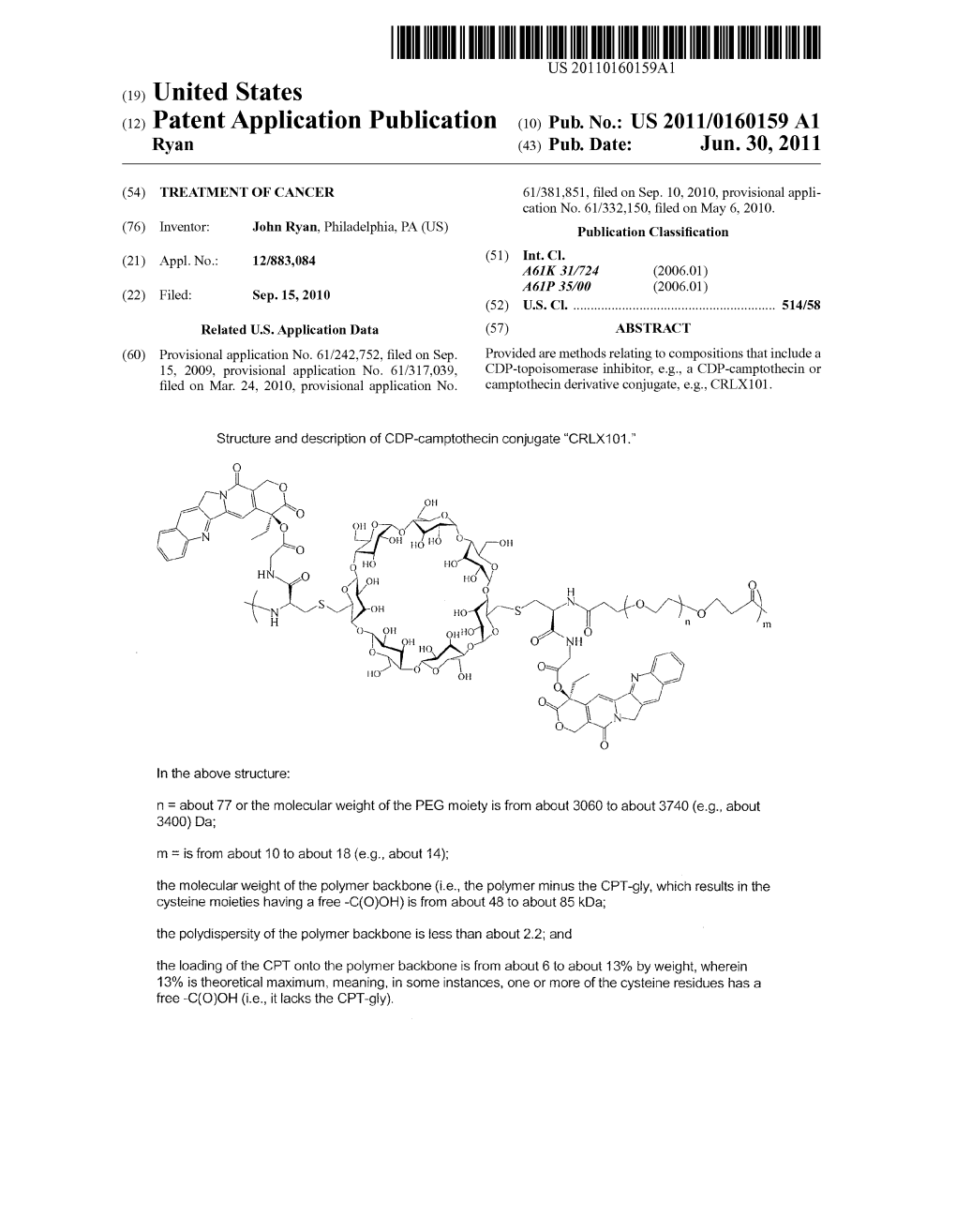
(12) Patent Application Publication (10) Pub. No.: US 2011/0160159 A1 Ryan (43) Pub
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WHO Drug Information Vol
WHO Drug Information Vol. 31, No. 3, 2017 WHO Drug Information Contents Medicines regulation 420 Post-market monitoring EMA platform gains trade mark; Automated 387 Regulatory systems in India FDA field alert reports 421 GMP compliance Indian manufacturers to submit self- WHO prequalification certification 421 Collaboration 402 Prequalification process quality China Food and Drug Administration improvement initiatives: 2010–2016 joins ICH; U.S.-EU cooperation in inspections; IGDRP, IPRF initiatives to join 422 Medicines labels Safety news Improved labelling in Australia 423 Under discussion 409 Safety warnings 425 Approved Brimonidine gel ; Lactose-containing L-glutamine ; Betrixaban ; C1 esterase injectable methylprednisolone inhibitor (human) ; Meropenem and ; Amoxicillin; Azithromycin ; Fluconazole, vaborbactam ; Delafloxacin ; Glecaprevir fosfluconazole ; DAAs and warfarin and pibrentasvir ; Sofosbuvir, velpatasvir ; Bendamustine ; Nivolumab ; Nivolumab, and voxilaprevir ; Cladribine ; Daunorubicin pembrolizumab ; Atezolizumab ; Ibrutinib and cytarabine ; Gemtuzumab ozogamicin ; Daclizumab ; Loxoprofen topical ; Enasidenib ; Neratinib ; Tivozanib ; preparations ; Denosumab ; Gabapentin Guselkumab ; Benznidazole ; Ciclosporin ; Hydroxocobalamine antidote kit paediatric eye drops ; Lutetium oxodotreotide 414 Diagnostics Gene cell therapy Hightop HIV home testing kits Tisagenlecleucel 414 Known risks Biosimilars Warfarin ; Local corticosteroids Bevacizumab; Adalimumab ; Hydroquinone skin lighteners Early access 415 Review outcomes Idebenone -

RESEARCH ARTICLE Lobaplatin Combined Floxuridine/Pirarubicin
DOI:http://dx.doi.org/10.7314/APJCP.2014.15.5.2057 The Effect Observation of LBP-based TACE for Unresectable Primary Hepatocellular Carcinoma RESEARCH ARTICLE Lobaplatin Combined Floxuridine/Pirarubicin-based Transcatheter Hepatic Arterial Chemoembolization for Unresectable Primary Hepatocellular Carcinoma Chang Zhao1, Xu-Jie Wang2*, Song Wang2*, Wei-Hua Feng2, Lei Shi2, Chun- Peng Yu2 Abstract Purpose: To assess the effect and safety of lobaplatin combinated floxuridine /pirarubicin in transcatheter hepatic arterial chemoembolization(TACE) of unresectable primary liver cancer. Patients and Methods: TACE combined with the chemotherapy regimen was used to treat 34 unresectable primary liver cancer patients. DSA/ MRI/CT/blood routine examinations were used to evaluate short term activity and toxicity after 4-5 weeks, the process being repeated if necessary. Results: Among the 34 cases, 1 (2.9%) showed a complete response, 21 (61.7%) a partial response, 8 (23.5%) stable disease, and 4 progressive disease, with a total effective rate of 67.6%. The content of alpha fetoprotein dropped by over 50% in 20 cases (58.8%). The rate of recovery was hepatalgia (88.2%), ascites (47.1%), appetite (55.9%), Performance Status(30.4%). The median follow-up time (MFT) was 281 days (63-558 days), and median progression-free survival was 118.5 days (95%, CI:88.8-148.2days). Adverse reactions (III-IV grade) were not common, with only 4 cases of vomiting and 2 cases of thrombocytopenia (III grade). Conclusions: Lobaplatin-based TACE is an effective and safe treatment for primary liver cancer. Keywords: Primary hepatic carcinoma - chemoembolization - platinum chemotherapy - clinical response Asian Pac J Cancer Prev, 15 (5), 2057-2060 Introduction LBP-based TACE, male 29 cases, female 5 cases, average age for 57.2±10.7 years, 12 cases were biopsy-proven, Hepatocellular carcinoma (HCC) is a common 22 cases were in line with the China Cancer Association malignant tumor . -

Camptothecin Derivatives Camptothecin-Derivate Dérivés De La Camptothécine
(19) TZZ ¥_ _T (11) EP 2 443 125 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07D 491/14 (2006.01) C07D 491/22 (2006.01) 26.11.2014 Bulletin 2014/48 A61P 35/00 (2006.01) A61K 31/4741 (2006.01) (21) Application number: 10790151.4 (86) International application number: PCT/US2010/038890 (22) Date of filing: 16.06.2010 (87) International publication number: WO 2010/148138 (23.12.2010 Gazette 2010/51) (54) CAMPTOTHECIN DERIVATIVES CAMPTOTHECIN-DERIVATE DÉRIVÉS DE LA CAMPTOTHÉCINE (84) Designated Contracting States: • CAO: "Preparationof 14- nitrocamptothecin...", J. AL AT BE BG CH CY CZ DE DK EE ES FI FR GB CHEM.SOC., PERKIN TRANS. 1, vol. 21, 1996, GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO pages 2629-2632, XP002684965, PL PT RO SE SI SK SM TR • CHENG KEJUN ET AL: "14-azacamptothecin: a potent water-soluble topoisomerase I poison.", (30) Priority: 17.06.2009 US 218043 P JOURNAL OF THE AMERICAN CHEMICAL 16.04.2010 US 325223 P SOCIETY 26 JAN 2005 LNKD- PUBMED: 15656613, vol. 127, no. 3, 26 January 2005 (43) Date of publication of application: (2005-01-26), pages838-839, XP002684966, ISSN: 25.04.2012 Bulletin 2012/17 0002-7863 • VADWAI VIRAL ET AL: "Insilico analysis of (73) Proprietor: Threshold Pharmaceuticals, Inc. homocamptothecin (hCPT) analogues for anti- Redwood City, California 94063 (US) tumour activity", INTERNATIONAL JOURNAL OF BIOINFORMATICS RESEARCH AND (72) Inventors: APPLICATIONS, INDERSCIENCE PUBLISHERS, • CAI, Xiaohong BUCKS, GB, vol. -

Weak Interaction of the Antimetabolite Drug Methotrexate with a Cavitand Derivative
International Journal of Molecular Sciences Article Weak Interaction of the Antimetabolite Drug Methotrexate with a Cavitand Derivative Zsolt Preisz 1,2, Zoltán Nagymihály 3,4, Beáta Lemli 1,4 ,László Kollár 3,4 and Sándor Kunsági-Máté 1,2,4,* 1 Institute of Organic and Medicinal Chemistry, Medical School, University of Pécs, Szigeti 12, H-7624 Pécs, Hungary; [email protected] (Z.P.); [email protected] (B.L.) 2 Department of General and Physical Chemistry, Faculty of Sciences, University of Pécs, Ifjúság 6, H 7624 Pécs, Hungary 3 Department of Inorganic Chemistry, Faculty of Sciences, University of Pécs, Ifjúság 6, H 7624 Pécs, Hungary; [email protected] (Z.N.); [email protected] (L.K.) 4 János Szentágothai Research Center, University of Pécs, Ifjúság 20, H-7624 Pécs, Hungary * Correspondence: [email protected]; Tel.: +36-72-503600 (ext. 35449) Received: 2 June 2020; Accepted: 16 June 2020; Published: 18 June 2020 Abstract: Formation of inclusion complexes involving a cavitand derivative (as host) and an antimetabolite drug, methotrexate (as guest) was investigated by photoluminescence measurements in dimethyl sulfoxide solvent. Molecular modeling performed in gas phase reflects that, due to the structural reasons, the cavitand can include the methotrexate in two forms: either by its opened structure with free androsta-4-en-3-one-17α-ethinyl arms or by the closed form when all the androsta-4-en-3-one-17α-ethinyl arms play role in the complex formation. Experiments reflect enthalpy driven complex formation in higher temperature range while at lower temperature the complexes are stabilized by the entropy gain. -

In Vitro and in Vivo Reversal of Resistance to 5-Fluorouracil in Colorectal Cancer Cells with a Novel Stealth Double-Liposomal Formulation
British Journal of Cancer (2007) 97, 919 – 926 & 2007 Cancer Research UK All rights reserved 0007 – 0920/07 $30.00 www.bjcancer.com In vitro and in vivo reversal of resistance to 5-fluorouracil in colorectal cancer cells with a novel stealth double-liposomal formulation 1 1 1 1 2 3 *,1 R Fanciullino , S Giacometti , C Mercier , C Aubert , C Blanquicett , P Piccerelle and J Ciccolini 1 2 EA3286-Laboratoire de Pharmacocine´tique, Universite´ de la Me´diterrane´e, Marseille, France; Department of Medicine, School of Medicine, Emory 3 University, Atlanta, GA, USA; Laboratoire de Pharmacie Gale´nique, Faculte´ de Pharmacie, 27 Bd Jean Moulin, Marseille 05 13385, France Drug resistance is a major cause of treatment failure in cancer chemotherapy, including that with the extensively prescribed antimetabolite, 5-fluorouracil (5-FU). In this study, we tried to reverse 5-FU resistance by using a double-punch strategy: combining 5-FU with a biochemical modulator to improve its tumoural activation and encapsulating both these agents in one same stealth liposome. Experiments carried out in the highly resistant, canonical SW620 human colorectal model showed a up to 80% sensitisation to 5-FU when these cells were treated with our liposomal formulation. Results with this formulation demonstrated 30% higher tumoural drug uptake, better activation with increased active metabolites including critical-5-fluoro-2-deoxyuridine-5-monophos- phate, superior inhibition (98%) of tumour thymidylate synthase, and subsequently, higher induction of both early and late apoptosis. Drug monitoring showed that higher and sustained exposure was achieved in rats treated with liposomal formulation. When examined in a xenograft animal model, our dual-agent liposomal formulation caused a 74% reduction in tumour size with a mean doubling in survival time, whereas standard 5-FU failed to exhibit significant antiproliferative activity as well as to increase the lifespan of tumour-bearing mice. -

Public Assessment Report
Public Assessment Report Tifix 150mg and 500mg film-coated tablets Capecitabine BE/H/195/01-02/DC BE licence no: BE423552-BE423561 Applicant: Alfred E. Tiefenbacher, Germany Date: 27-04-2012 Toepassingsdatum : 15-09-10 Page 1 of 17 Blz. 1 van 17 This assessment report is published by the Federal Agency for Medicines and Health Products following Article 21 (3) and (4) of Directive 2001/83/EC, amended by Directive 2004/27/EC and Article 25 paragraph 4 of Directive 2001/82/EC as amended by 2004/28/EC. The report comments on the registration dossier that was submitted to the Federal Agency for Medicines and Health Products and its fellow organisations in all concerned EU member states. It reflects the scientific conclusion reached by the Federal Agency for Medicines and Health Products and all concerned member states at the end of the evaluation process and provides a summary of the grounds for approval of a marketing authorisation. This report is intended for all those involved with the safe and proper use of the medicinal product, i.e. healthcare professionals, patients and their family and carers. Some knowledge of medicines and diseases is expected of the latter category as the language in this report may be difficult for laymen to understand. This assessment report shall be updated by a following addendum whenever new information becomes available. To the best of the Federal Agency for Medicines and Health Products’ knowledge, this report does not contain any information that should not have been made available to the public. The Marketing Autorisation Holder has checked this report for the absence of any confidential information. -

Hepatic Arterial Infusion for Unresectable Colorectal Liver Metastases Combined Or Not with Systemic Chemotherapy
ANTICANCER RESEARCH 29: 4139-4144 (2009) Hepatic Arterial Infusion for Unresectable Colorectal Liver Metastases Combined or Not with Systemic Chemotherapy PIERLUIGI PILATI, ENZO MAMMANO, SIMONE MOCELLIN, EMANUELA TESSARI, MARIO LISE and DONATO NITTI Clinica Chirurgica II, Department of Oncological and Surgical Sciences, University of Padova, 35128 Padova, Italy Abstract. Background: The hypothesis was tested that CRC have liver metastases at autopsy, and the majority of systemic chemotherapy might contribute to improving overall these patients die as a result of their metastatic disease. survival (OS) of patients with unresectable colorectal liver Surgical resection represents the standard treatment of metastases treated with hepatic arterial infusion (HAI). resectable disease and is followed by 5-year overall Patients and Methods: We considered 153 consecutive patients survival (OS) rates of 20% to 40% (2, 3). Unfortunately, retrospectively divided into group A (n=72) treated with HAI only 20% of patients with liver metastases from CRC alone (floxuridine [FUDR] + leucovorin [LV]), and group B present with liver-confined resectable disease and/or are (n=81) treated with HAI combined with systemic candidates for major surgical operation (depending on chemotherapy (5-fluorouracil [5FU] + LV). Results: No comorbidities) (4, 5). significant difference in OS was observed between the two The therapeutic management of unresectable metastases is groups. Median OS was better in patients with <50% of liver more controversial and is generally associated with a dismal involvement (21.3 vs. 13.2 months; p<0.0001) and in prognosis. In fact, despite the improvements achieved with responders vs. non-responders (24.4 vs. 13.4 months; modern systemic chemotherapy (SCT) regimens (e.g. -

Evaluation of Floxuridine Oligonucleotide Conjugates Carrying Potential Enhancers of Cellular Uptake
International Journal of Molecular Sciences Article Evaluation of Floxuridine Oligonucleotide Conjugates Carrying Potential Enhancers of Cellular Uptake Anna Aviñó 1,2,*, Anna Clua 1,2 , Maria José Bleda 1 , Ramon Eritja 1,2,* and Carme Fàbrega 1,2 1 Institute for Advanced Chemistry of Catalonia (IQAC-CSIC), Jordi Girona 18-26, E-08034 Barcelona, Spain; [email protected] (A.C.); [email protected] (M.J.B.); [email protected] (C.F.) 2 Networking Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Jordi Girona 18-26, E-08034 Barcelona, Spain * Correspondence: [email protected] (A.A.); [email protected] (R.E.); Tel.: +34-934-006-100 (A.A.) Abstract: Conjugation of small molecules such as lipids or receptor ligands to anti-cancer drugs has been used to improve their pharmacological properties. In this work, we studied the biological effects of several small-molecule enhancers into a short oligonucleotide made of five floxuridine units. Specifically, we studied adding cholesterol, palmitic acid, polyethyleneglycol (PEG 1000), folic acid and triantennary N-acetylgalactosamine (GalNAc) as potential enhancers of cellular uptake. As expected, all these molecules increased the internalization efficiency with different degrees depending on the cell line. The conjugates showed antiproliferative activity due to their metabolic activation by nuclease degradation generating floxuridine monophosphate. The cytotoxicity and apoptosis assays showed an increase in the anti-cancer activity of the conjugates related to the floxuridine oligomer, but this effect did not correlate with the internalization results. Palmitic and folic acid conjugates provide the highest antiproliferative activity without having the highest internalization Citation: Aviñó, A.; Clua, A.; Bleda, results. -

Drugs and Life-Threatening Ventricular Arrhythmia Risk: Results from the DARE Study Cohort
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016627 on 16 October 2017. Downloaded from Drugs and life-threatening ventricular arrhythmia risk: results from the DARE study cohort Abigail L Coughtrie,1,2 Elijah R Behr,3,4 Deborah Layton,1,2 Vanessa Marshall,1 A John Camm,3,4,5 Saad A W Shakir1,2 To cite: Coughtrie AL, Behr ER, ABSTRACT Strengths and limitations of this study Layton D, et al. Drugs and Objectives To establish a unique sample of proarrhythmia life-threatening ventricular cases, determine the characteristics of cases and estimate ► The Drug-induced Arrhythmia Risk Evaluation study arrhythmia risk: results from the the contribution of individual drugs to the incidence of DARE study cohort. BMJ Open has allowed the development of a cohort of cases of proarrhythmia within these cases. 2017;7:e016627. doi:10.1136/ proarrhythmia. Setting Suspected proarrhythmia cases were referred bmjopen-2017-016627 ► These cases have provided crucial safety by cardiologists across England between 2003 and 2011. information, as well as underlying clinical and ► Prepublication history for Information on demography, symptoms, prior medical and genetic data. this paper is available online. drug histories and data from hospital notes were collected. ► Only patients who did not die as a result of the To view these files please visit Participants Two expert cardiologists reviewed data the journal online (http:// dx. doi. proarrhythmia could be included. for 293 referred cases: 130 were included. Inclusion org/ 10. 1136/ bmjopen- 2017- ► Referral of cases by cardiologists alone may have criteria were new onset or exacerbation of pre-existing 016627). -

Polymer-Drug Conjugate, a Potential Therapeutic to Combat Breast and Lung Cancer
pharmaceutics Review Polymer-Drug Conjugate, a Potential Therapeutic to Combat Breast and Lung Cancer Sibusiso Alven, Xhamla Nqoro , Buhle Buyana and Blessing A. Aderibigbe * Department of Chemistry, University of Fort Hare, Alice Eastern Cape 5700, South Africa; [email protected] (S.A.); [email protected] (X.N.); [email protected] (B.B.) * Correspondence: [email protected] Received: 24 November 2019; Accepted: 30 December 2019; Published: 29 April 2020 Abstract: Cancer is a chronic disease that is responsible for the high death rate, globally. The administration of anticancer drugs is one crucial approach that is employed for the treatment of cancer, although its therapeutic status is not presently satisfactory. The anticancer drugs are limited pharmacologically, resulting from the serious side effects, which could be life-threatening. Polymer drug conjugates, nano-based drug delivery systems can be utilized to protect normal body tissues from the adverse side effects of anticancer drugs and also to overcome drug resistance. They transport therapeutic agents to the target cell/tissue. This review article is based on the therapeutic outcomes of polymer-drug conjugates against breast and lung cancer. Keywords: breast cancer; lung cancer; chemotherapy; polymer-based carriers; polymer-drug conjugates 1. Introduction Cancer is a chronic disease that leads to great mortality around the world and cancer cases are rising continuously [1]. It is the second cause of death worldwide, followed by cardiovascular diseases [2]. It is characterized by an abnormal uncontrolled proliferation of any type of cells in the human body [3]. It is caused by external factors, such as smoking, infectious organisms, pollution, and radiation; it is also caused by internal factors, such as immune conditions, hormones, and genetic mutation [3]. -

Recent Advances in Arsenic Trioxide Encapsulated Nanoparticles As Drug Delivery Agents to Solid Cancers
Available online at www.jbr-pub.org Open Access at PubMed Central The Journal of Biomedical Research, 2017 31(3): 177–188 Review Article Recent advances in arsenic trioxide encapsulated nanoparticles as drug delivery agents to solid cancers Anam Akhtar, Scarlet Xiaoyan Wang, Lucy Ghali, Celia Bell, Xuesong Wen✉ Department of Natural Sciences, School of Science and Technology, Middlesex University, London NW4 4BT, UK. Abstract Since arsenic trioxide was first approved as the front line therapy for acute promyelocytic leukemia 25 years ago, its anti-cancer properties for various malignancies have been under intense investigation. However, the clinical successes of arsenic trioxide in treating hematological cancers have not been translated to solid cancers. This is due to arsenic's rapid clearance by the body's immune system before reaching the tumor site. Several attempts have henceforth been made to increase its bioavailability toward solid cancers without increasing its dosage albeit without much success. This review summarizes the past and current utilization of arsenic trioxide in the medical field with primary focus on the implementation of nanotechnology for arsenic trioxide delivery to solid cancer cells. Different approaches that have been employed to increase arsenic's efficacy, specificity and bioavailability to solid cancer cells were evaluated and compared. The potential of combining different approaches or tailoring delivery vehicles to target specific types of solid cancers according to individual cancer characteristics and arsenic chemistry is proposed and discussed. Keywords: arsenic trioxide, solid cancer, nanotechnology, drug delivery, liposome Introduction Despite being the mainstay of materia medica since the past 2,400 years, ATO faced an escalating decline in Arsenic is a naturally occurring metalloid found as a its use as a therapeutic agent in the twentieth century [1] trace element in the earth's crust . -

Palladium-Mediated Dealkylation of N-Propargyl-Floxuridine As a Bioorthogonal Oxygen-Independent Prodrug Strategy
OPEN Palladium-Mediated Dealkylation of SUBJECT AREAS: N-Propargyl-Floxuridine as a CHEMOTHERAPY CHEMICAL TOOLS Bioorthogonal Oxygen-Independent DRUG DISCOVERY AND DEVELOPMENT Prodrug Strategy Jason T. Weiss, Neil O. Carragher & Asier Unciti-Broceta Received 2 December 2014 Edinburgh Cancer Research UK Centre, MRC Institute of Genetics and Molecular Medicine, University of Edinburgh, Crewe Road Accepted South, Edinburgh EH4 2XR, UK. 26 February 2015 Published 19 March 2015 Herein we report the development and biological screening of a bioorthogonal palladium-labile prodrug of the nucleoside analogue floxuridine, a potent antineoplastic drug used in the clinic to treat advanced cancers. N-propargylation of the N3 position of its uracil ring resulted in a vast reduction of its biological activity (,6,250-fold). Cytotoxic properties were bioorthogonally rescued in cancer cell culture by Correspondence and heterogeneous palladium chemistry both in normoxia and hypoxia. Within the same environment, the requests for materials reported chemo-reversible prodrug exhibited up to 1,450-fold difference of cytotoxicity whether it was in the absence or presence of the extracellular palladium source, underlining the precise modulation of bioactivity should be addressed to enabled by this bioorthogonally-activated prodrug strategy. A.U.-B. (Asier.Unciti- [email protected]. uk) ioorthogonally-activated prodrug therapies are a heterogeneous group of experimentally and clinically- used therapeutic strategies that are founded on a common principle: the site-specific activation of phar- B maceutical substances by the mediation of non-biological, non-perturbing physical or chemical stimuli. While the nature and properties of the triggering stimulus can be manifestly diverse and seemingly unrelated (e.g.