The Role of Fc Receptor-Like 6 in Innate and Adaptive Immunity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Ligands for Human Igg and Their Effector Functions
antibodies Review The Ligands for Human IgG and Their Effector Functions Steven W. de Taeye 1,2,*, Theo Rispens 1 and Gestur Vidarsson 2 1 Sanquin Research, Dept Immunopathology and Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam, 1066 CX Amsterdam, The Netherlands; [email protected] 2 Sanquin Research, Dept Experimental Immunohematology and Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam, 1066 CX Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected] Received: 26 March 2019; Accepted: 18 April 2019; Published: 25 April 2019 Abstract: Activation of the humoral immune system is initiated when antibodies recognize an antigen and trigger effector functions through the interaction with Fc engaging molecules. The most abundant immunoglobulin isotype in serum is Immunoglobulin G (IgG), which is involved in many humoral immune responses, strongly interacting with effector molecules. The IgG subclass, allotype, and glycosylation pattern, among other factors, determine the interaction strength of the IgG-Fc domain with these Fc engaging molecules, and thereby the potential strength of their effector potential. The molecules responsible for the effector phase include the classical IgG-Fc receptors (FcγR), the neonatal Fc-receptor (FcRn), the Tripartite motif-containing protein 21 (TRIM21), the first component of the classical complement cascade (C1), and possibly, the Fc-receptor-like receptors (FcRL4/5). Here we provide an overview of the interactions of IgG with effector molecules and discuss how natural variation on the antibody and effector molecule side shapes the biological activities of antibodies. The increasing knowledge on the Fc-mediated effector functions of antibodies drives the development of better therapeutic antibodies for cancer immunotherapy or treatment of autoimmune diseases. -

Annual Report 2015
McGill University Department of Microbiology and Immunology 2015 Annual Report Submitted by J. Madrenas, MD, PhD, FCAHS Canada Research Chair in Human Immunology Professor and Chairman On behalf of Primary members of the Department of Microbiology and Immunology Section I Introduction A. Executive Summary 2015 was an exciting year in the life of the Department of Microbiology and Immunology at McGill. During 2015, we had an External Cyclical Review of the Department, the first review of its kind for (at least) the last 22 years. This process identified key strengths and challenges for our unit, and coalesced different strategic initiatives in consultation with the leadership in the Faculty of Medicine and the Provost office. In 2015, the department welcomed Dr. Corinne Maurice arriving from Harvard University. Corinne started as a tenure track assistant professor on January 1. Her arrival was the fourth recruitment in the context of departmental faculty renewal in the last three years. In addition, we completed the selection process to recruit a Virologist to the Department: the selected candidate was Dr. Jacques Archambault, who will be joining on August 1, 2016 as a Full Professor with tenure. We successfully completed the promotion to Full Professors of Drs. Ciriacco Piccirillo and Donald Sheppard and the 3 yr. reappointment process for Dr. Selena Sagan, and initiated the promotion to Associate Professor with tenure for Dr. Jorg Fritz (a process successfully completed in May 2016). In Research, our members were very successful in attracting high calibre awards. Dr. Mark Wainberg was a 2016 Canadian Medical Hall of Fame inductee. Dr. -

Genetic Variations in the PSMA6 and PSMC6 Proteasome Genes Are Associated with Multiple Sclerosis and Response to Interferon‑Β Therapy in Latvians
EXPERIMENTAL AND THERAPEUTIC MEDICINE 21: 478, 2021 Genetic variations in the PSMA6 and PSMC6 proteasome genes are associated with multiple sclerosis and response to interferon‑β therapy in Latvians NATALIA PARAMONOVA1, JOLANTA KALNINA1, KRISTINE DOKANE1, KRISTINE DISLERE1, ILVA TRAPINA1, TATJANA SJAKSTE1 and NIKOLAJS SJAKSTE1,2 1Genomics and Bioinformatics, Institute of Biology of The University of Latvia; 2Department of Medical Biochemistry of The University of Latvia, LV‑1004 Riga, Latvia Received July 8, 2020; Accepted December 8, 2020 DOI: 10.3892/etm.2021.9909 Abstract. Several polymorphisms in genes related to the Introduction ubiquitin‑proteasome system exhibit an association with pathogenesis and prognosis of various human autoimmune Multiple sclerosis (MS) is a lifelong demyelinating disease of diseases. Our previous study reported the association the central nervous system. The clinical onset of MS tends to between multiple sclerosis (MS) and the PSMA3‑rs2348071 be between the second and fourth decade of life. Similarly to polymorphism in the Latvian population. The current study other autoimmune diseases, women are affected 3‑4 times more aimed to evaluate the PSMA6 and PSMC6 genetic variations, frequently than men (1). About 10% of MS patients experience their interaction between each other and with the rs2348071, a primary progressive MS form characterized by the progres‑ on the susceptibility to MS risk and response to therapy in sion of neurological disability from the onset. In about 90% the Latvian population. PSMA6‑rs2277460, ‑rs1048990 and of MS patients, the disease undergoes the relapse‑remitting PSMC6‑rs2295826, ‑rs2295827 were genotyped in the MS MS course (RRMS); in most of these patients, the condition case/control study and analysed in terms of genotype‑protein acquires secondary progressive course (SPMS) (2). -

This Is an Author Produced Version of a Paper Published in Immunology
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Publications from Karolinska Institutet This is an author produced version of a paper published in Immunology. This paper has been peer-reviewed but does not include the final publisher proof-corrections or journal pagination. Pubmed citation for the paper: Immunology. 2014 May 6. [Epub ahead of print] Ligation of human Fc receptor like-2 (FCRL2) by monoclonal antibodies downregulates B cell receptor mediated signaling. Shabani M, Bayat AA, Jeddi-Tehrani M, Rabbani H, Hojjat-Farsangi M, Ulivieri C, Amirghofran Z, Baldari CT, Shokri F. URL: http://dx.doi.org/10.1111/imm.12311 Access to the published version may require subscription. Published with permission from: Wiley Ligation of human Fc receptor like-2 (FCRL2) by monoclonal antibodies downregulates B cell receptor mediated signaling Mahdi Shabania,b, Ali Ahmad Bayata, Mahmood Jeddi-Tehrania, Hodjatallah Rabbania,c, Mohammad Hojjat-Farsangid, Cristina Ulivierie, Zahra Amirghofranb, Cosima Tatiana Baldarie and Fazel Shokria,f* a Monoclonal Antibody Research Center, Avicenna Research Institute, ACECR, Tehran, Iran b Department of Immunology, Medical School, Shiraz University of Medical Sciences, Shiraz, Iran c Division of Clinical Immunology, Department of Laboratory Medicine, Karolinska Institutet at Karolinska University Hospital Huddinge, SE-14186 Stockholm, Sweden d Immune and Gene Therapy Laboratory, Cancer Centre Karolinska, Department of Oncology-Pathology, Karolinska Institutet, Stockholm e Department of Life Sciences, University of Siena, Siena, Italy f Department of Immunology, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran *Corresponding author: Fazel Shokri, Ph.D., Professor, Monoclonal Antibody Research Center, Avicenna Research Institute, ACECR, Tehran, Iran, P.O. -

Fcrl5 and T-Bet Define Influenza-Specific Memory B Cells That Predict Long-Lived Antibody 2 Responses 3 4 Anoma Nellore1, Christopher D
bioRxiv preprint doi: https://doi.org/10.1101/643973; this version posted May 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Fcrl5 and T-bet define influenza-specific memory B cells that predict long-lived antibody 2 responses 3 4 Anoma Nellore1, Christopher D. Scharer2, Rodney G. King3, Christopher M. Tipton4, Esther 5 Zumaquero3, Christopher Fucile3,5, Betty Mousseau3, John E. Bradley6, Kevin Macon3, Tian Mi2, 6 Paul A. Goepfert1,3, John F. Kearney,3 Jeremy M. Boss,2 Troy D. Randall6, Ignacio Sanz4, 7 Alexander Rosenberg3,5, Frances E. Lund3 8 9 10 11 1Dept. of Medicine, Division of Infectious Disease, 3Dept. of Microbiology, 5Informatics Institute, 12 6Dept of Medicine, Division of Clinical Immunology and Rheumatology, at The University of 13 Alabama at Birmingham, Birmingham, AL 35294 USA 14 15 2Dept. of Microbiology and Immunology and 4Department of Medicine, Division of 16 Rheumatology, at Emory University, Atlanta, GA 30322, USA 17 18 19 20 21 22 23 24 25 26 27 28 Lead Contact and to whom correspondence should be addressed: [email protected] 29 30 Mailing address: Frances E. Lund, PhD 31 Charles H. McCauley Professor and Chair 32 Dept of Microbiology 33 University of Alabama at Birmingham (UAB) 34 Dept of Microbiology 35 University of Alabama at Birmingham (UAB) 36 276 BBRB Box 11 37 1720 2nd Avenue South 38 Birmingham AL 35294-2170 39 40 SHORT RUNNING TITLE: Effector memory B cell development after influenza vaccination 41 bioRxiv preprint doi: https://doi.org/10.1101/643973; this version posted May 20, 2019. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Rabbit Anti-Phospho-PTPN6-SL5578R-FITC
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-phospho-PTPN6 SL5578R-FITC Product Name: Anti-phospho-PTPN6(Ser591)/FITC Chinese Name: FITC标记的磷酸化蛋白酪氨酸磷酸酶1C抗体 PTPN6(phospho S591); SHP1(phospho S591); p-SHP1(Ser591); SHP-1(Phospho- Tyr591); PTPN6(phospho Y591); PTPN6(phospho Y591); SHP1; 70 kda SHP 1L protein; 70 kda SHP1L protein; 70Z-SHP; EC 3.1.3.48; HCP; HCPH; Hematopoietic cell phosphatase; Hematopoietic cell protein tyrosine phosphatase; Hematopoietic cell protein-tyrosine phosphatase; HPTP 1C; HPTP1C; MGC124580; Protein tyrosine Alias: phosphatase 1C; Protein tyrosine phosphatase non receptor type 6; Protein tyrosine phosphatase SHP 1; Protein tyrosine phosphatase SHP1; Protein-tyrosine phosphatase 1C; Protein-tyrosine phosphatase SHP-1; PTN6_HUMAN; PTP 1C; PTP-1C; PTP1C; PTPN 6; PTPN6; SH PTP 1; SH PTP1; SH-PTP1; SHP 1; SHP 1L; SHP1L; SHPTP 1; SHPTP1; Tyrosine protein phosphatase non receptor type 6; Tyrosine-protein phosphatase non-receptor type 6. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Dog,Pig,Horse, Flow-Cyt=1:50-200IF=1:50-200www.sunlongbiotech.com Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 65kDa Form: Lyophilized or Liquid Concentration: 1mg/ml KLH conjugated Synthesised phosphopeptide derived from human PTPN6 around the immunogen: phosphorylation site of Ser591 [KG(p-S)LK] Lsotype: IgG Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Characteristics of B Cell-Associated Gene Expression in Patients With
MOLECULAR MEDICINE REPORTS 13: 4113-4121, 2016 Characteristics of B cell-associated gene expression in patients with coronary artery disease WENWEN YAN*, HAOMING SONG*, JINFA JIANG, WENJUN XU, ZHU GONG, QIANGLIN DUAN, CHUANGRONG LI, YUAN XIE and LEMIN WANG Department of Internal Medicine, Division of Cardiology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, P.R. China Received May 19, 2015; Accepted February 12, 2016 DOI: 10.3892/mmr.2016.5029 Abstract. The current study aimed to identify differentially with the two other groups. Additionally the gene expression expressed B cell-associated genes in peripheral blood mono- levels of B cell regulatory genes were measured. In patients nuclear cells and observe the changes in B cell activation at with AMI, CR1, LILRB2, LILRB3 and VAV1 mRNA expres- different stages of coronary artery disease. Groups of patients sion levels were statistically increased, whereas, CS1 and IL4I1 with acute myocardial infarction (AMI) and stable angina (SA), mRNAs were significantly reduced compared with the SA and as well as healthy volunteers, were recruited into the study control groups. There was no statistically significant difference (n=20 per group). Whole human genome microarray analysis in B cell-associated gene expression levels between patients was performed to examine the expression of B cell-associated with SA and the control group. The present study identified the genes among these three groups. The mRNA expression levels downregulation of genes associated with BCRs, B2 cells and of 60 genes associated with B cell activity and regulation were B cell regulators in patients with AMI, indicating a weakened measured using reverse transcription-quantitative polymerase T cell-B cell interaction and reduced B2 cell activation during chain reaction. -

Disease Phenotypes and Gender Association of FCRL3 Single
Disease Phenotypes and Gender Association of FCRL3 Single-Nucleotide Polymorphism –169T/C in Taiwanese Patients with Systemic Lupus Erythematosus and Rheumatoid Arthritis JI-YIH CHEN, CHIN-MAN WANG, YEONG-JIAN JAN WU, SHIN-NING KUO, CHIUNG-FANG SHIU, SU-WEI CHANG, YEN-TSUN LIN, HUEI-HUANG HO, and JIANMING WU ABSTRACT. Objective. To investigate the association of the functional FCRL3 single-nucleotide polymorphism (SNP) –169T/C with disease phenotypes and susceptibility to systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) in Taiwanese. Methods. FCRL3 SNP –169T/C was genotyped in 573 patients with SLE, 670 patients with RA, and 758 controls. Genotype distributions and allele frequencies were compared among the 3 groups as aggregates or as stratified by clinical characteristics, autoantibody profile, and sex within patient groups. Results. Overall, FCRL3 SNP –169T/C was not associated with susceptibility to either SLE or RA. However, –169CC genotype was significantly reduced in leukopenia-positive SLE patients as com- pared to the leukopenia-negative SLE patients (CC vs CT+TT, p = 6 × 10–4, OR 0.444, 95% CI 0.279–0.708) and controls (p = 6.1 × 10–3, OR 0.583, 95% CI 0.396–0.857). On the other hand, –169TT genotypes were significantly more numerous in RA patients with non-destructive disease as compared with patients with destructive disease (CC+CT vs TT: p = 0.007, OR 1.672, 95% CI 1.149–2.432). The –169T allele frequency was also significantly increased in non-destructive RA compared with patients with destructive disease (C vs T: p = 0.010, OR 1.423, 95% CI 1.089–1.859). -
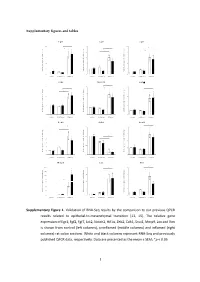
Type of the Paper (Article
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Monoclonal Antibody-Based Therapy As a New Treatment Strategy in Multiple Myeloma
Leukemia (2012) 26, 199–213 & 2012 Macmillan Publishers Limited All rights reserved 0887-6924/12 www.nature.com/leu REVIEW Monoclonal antibody-based therapy as a new treatment strategy in multiple myeloma NWCJ van de Donk1, S Kamps1, T Mutis2 and HM Lokhorst1 1Department of Hematology, University Medical Center Utrecht, Utrecht, The Netherlands and 2Department of Clinical Chemistry and Hematology, University Medical Center Utrecht, Utrecht, The Netherlands The introduction of autologous stem cell transplantation the myeloma patients achieved a partial response (PR) or stable combined with the introduction of immunomodulatory drugs disease following rituximab therapy. All these patients expressed (IMiDs) and proteasome inhibitors has significantly improved CD20 on their myeloma cells.2 However, as only B15–20% of survival of multiple myeloma patients. However, ultimately the majority of patients will develop refractory disease, indicating all myeloma patients express CD20 on their bone marrow the need for new treatment modalities. In preclinical and clinical plasma cells, new targets for immunotherapy need to be studies, promising results have been obtained with several identified. The search for other targets has led to the develop- monoclonal antibodies (mAbs) targeting the myeloma tumor ment of mAbs targeting growth factor receptors or adhesion cell or the bone marrow microenvironment. The mechanisms molecules on myeloma cells. Other newly developed mAbs underlying the therapeutic efficacy of these mAbs include are directed against cellular or non-cellular components of the direct induction of tumor cell apoptosis via inhibition or activation of target molecules, complement-dependent cyto- bone marrow microenvironment, resulting in the neutraliza- toxicity and antibody-dependent cell-mediated cytotoxicity tion of growth factors, inhibition of angiogenesis, modulation (ADCC).