Letter to FDA on Review of Biogen's Drug
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Refreshing the Biologic Pipeline 2020
news feature Credit: Science Lab / Alamy Stock Photo Refreshing the biologic pipeline 2020 In the absence of face-to-face meetings, FDA and industry implemented regulatory workarounds to maintain drug and biologics approvals. These could be here to stay. John Hodgson OVID-19 might have been expected since 1996) — a small miracle in itself “COVID-19 confronted us with the need to severely impair drug approvals (Fig. 1 and Table 1). to better triage sponsors’ questions,” says Cin 2020. In the event, however, To the usual crop of rare disease and Peter Marks, the director of the Center for industry and regulators delivered a small genetic-niche cancer treatments, 2020 Biologics Evaluation and Research (CBER) miracle. They found workarounds and also added a chimeric antigen receptor at the FDA. “That was perhaps the single surrogate methods of engagement. Starting (CAR)-T cell therapy with a cleaner biggest takeaway from the pandemic related in January 2020, when the outbreak veered manufacturing process and the first to product applications.” Marks says that it westward, the number of face-to face approved blockbuster indication for a became very apparent with some COVID- meetings declined rapidly; by March, small-interfering RNA (siRNA) — the 19-related files that resolving a single they were replaced by Webex and Teams. European Medicines Agency’s (EMA) issue can help a sponsor enormously and (Secure Zoom meeting are to be added registration of the RNA interference accelerate the development cycle. Before this year.) And remarkably, by 31 December, (RNAi) therapy Leqvio (inclisiran) for COVID-19, it was conceivable that a small the US Food and Drug Administration cardiovascular disease. -

Horizon Scanning Status Report June 2019
Statement of Funding and Purpose This report incorporates data collected during implementation of the Patient-Centered Outcomes Research Institute (PCORI) Health Care Horizon Scanning System, operated by ECRI Institute under contract to PCORI, Washington, DC (Contract No. MSA-HORIZSCAN-ECRI-ENG- 2018.7.12). The findings and conclusions in this document are those of the authors, who are responsible for its content. No statement in this report should be construed as an official position of PCORI. An intervention that potentially meets inclusion criteria might not appear in this report simply because the horizon scanning system has not yet detected it or it does not yet meet inclusion criteria outlined in the PCORI Health Care Horizon Scanning System: Horizon Scanning Protocol and Operations Manual. Inclusion or absence of interventions in the horizon scanning reports will change over time as new information is collected; therefore, inclusion or absence should not be construed as either an endorsement or rejection of specific interventions. A representative from PCORI served as a contracting officer’s technical representative and provided input during the implementation of the horizon scanning system. PCORI does not directly participate in horizon scanning or assessing leads or topics and did not provide opinions regarding potential impact of interventions. Financial Disclosure Statement None of the individuals compiling this information have any affiliations or financial involvement that conflicts with the material presented in this report. Public Domain Notice This document is in the public domain and may be used and reprinted without special permission. Citation of the source is appreciated. All statements, findings, and conclusions in this publication are solely those of the authors and do not necessarily represent the views of the Patient-Centered Outcomes Research Institute (PCORI) or its Board of Governors. -
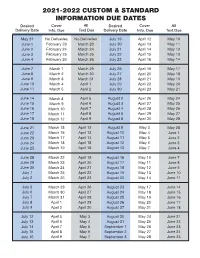
2021-2022 Custom & Standard Information Due Dates
2021-2022 CUSTOM & STANDARD INFORMATION DUE DATES Desired Cover All Desired Cover All Delivery Date Info. Due Text Due Delivery Date Info. Due Text Due May 31 No Deliveries No Deliveries July 19 April 12 May 10 June 1 February 23 March 23 July 20 April 13 May 11 June 2 February 24 March 24 July 21 April 14 May 12 June 3 February 25 March 25 July 22 April 15 May 13 June 4 February 26 March 26 July 23 April 16 May 14 June 7 March 1 March 29 July 26 April 19 May 17 June 8 March 2 March 30 July 27 April 20 May 18 June 9 March 3 March 31 July 28 April 21 May 19 June 10 March 4 April 1 July 29 April 22 May 20 June 11 March 5 April 2 July 30 April 23 May 21 June 14 March 8 April 5 August 2 April 26 May 24 June 15 March 9 April 6 August 3 April 27 May 25 June 16 March 10 April 7 August 4 April 28 May 26 June 17 March 11 April 8 August 5 April 29 May 27 June 18 March 12 April 9 August 6 April 30 May 28 June 21 March 15 April 12 August 9 May 3 May 28 June 22 March 16 April 13 August 10 May 4 June 1 June 23 March 17 April 14 August 11 May 5 June 2 June 24 March 18 April 15 August 12 May 6 June 3 June 25 March 19 April 16 August 13 May 7 June 4 June 28 March 22 April 19 August 16 May 10 June 7 June 29 March 23 April 20 August 17 May 11 June 8 June 30 March 24 April 21 August 18 May 12 June 9 July 1 March 25 April 22 August 19 May 13 June 10 July 2 March 26 April 23 August 20 May 14 June 11 July 5 March 29 April 26 August 23 May 17 June 14 July 6 March 30 April 27 August 24 May 18 June 15 July 7 March 31 April 28 August 25 May 19 June 16 July 8 April 1 April 29 August 26 May 20 June 17 July 9 April 2 April 30 August 27 May 21 June 18 July 12 April 5 May 3 August 30 May 24 June 21 July 13 April 6 May 4 August 31 May 25 June 22 July 14 April 7 May 5 September 1 May 26 June 23 July 15 April 8 May 6 September 2 May 27 June 24 July 16 April 9 May 7 September 3 May 28 June 25. -

Treatment of Alzheimer's Disease and Blood–Brain Barrier Drug Delivery
pharmaceuticals Review Treatment of Alzheimer’s Disease and Blood–Brain Barrier Drug Delivery William M. Pardridge Department of Medicine, University of California, Los Angeles, CA 90024, USA; [email protected] Received: 24 October 2020; Accepted: 13 November 2020; Published: 16 November 2020 Abstract: Despite the enormity of the societal and health burdens caused by Alzheimer’s disease (AD), there have been no FDA approvals for new therapeutics for AD since 2003. This profound lack of progress in treatment of AD is due to dual problems, both related to the blood–brain barrier (BBB). First, 98% of small molecule drugs do not cross the BBB, and ~100% of biologic drugs do not cross the BBB, so BBB drug delivery technology is needed in AD drug development. Second, the pharmaceutical industry has not developed BBB drug delivery technology, which would enable industry to invent new therapeutics for AD that actually penetrate into brain parenchyma from blood. In 2020, less than 1% of all AD drug development projects use a BBB drug delivery technology. The pathogenesis of AD involves chronic neuro-inflammation, the progressive deposition of insoluble amyloid-beta or tau aggregates, and neural degeneration. New drugs that both attack these multiple sites in AD, and that have been coupled with BBB drug delivery technology, can lead to new and effective treatments of this serious disorder. Keywords: blood–brain barrier; brain drug delivery; drug targeting; endothelium; Alzheimer’s disease; therapeutic antibodies; neurotrophins; TNF inhibitors 1. Introduction Alzheimer’s Disease (AD) afflicts over 50 million people world-wide, and this health burden costs over 1% of global GDP [1]. -

2021 7 Day Working Days Calendar
2021 7 Day Working Days Calendar The Working Day Calendar is used to compute the estimated completion date of a contract. To use the calendar, find the start date of the contract, add the working days to the number of the calendar date (a number from 1 to 1000), and subtract 1, find that calculated number in the calendar and that will be the completion date of the contract Date Number of the Calendar Date Friday, January 1, 2021 133 Saturday, January 2, 2021 134 Sunday, January 3, 2021 135 Monday, January 4, 2021 136 Tuesday, January 5, 2021 137 Wednesday, January 6, 2021 138 Thursday, January 7, 2021 139 Friday, January 8, 2021 140 Saturday, January 9, 2021 141 Sunday, January 10, 2021 142 Monday, January 11, 2021 143 Tuesday, January 12, 2021 144 Wednesday, January 13, 2021 145 Thursday, January 14, 2021 146 Friday, January 15, 2021 147 Saturday, January 16, 2021 148 Sunday, January 17, 2021 149 Monday, January 18, 2021 150 Tuesday, January 19, 2021 151 Wednesday, January 20, 2021 152 Thursday, January 21, 2021 153 Friday, January 22, 2021 154 Saturday, January 23, 2021 155 Sunday, January 24, 2021 156 Monday, January 25, 2021 157 Tuesday, January 26, 2021 158 Wednesday, January 27, 2021 159 Thursday, January 28, 2021 160 Friday, January 29, 2021 161 Saturday, January 30, 2021 162 Sunday, January 31, 2021 163 Monday, February 1, 2021 164 Tuesday, February 2, 2021 165 Wednesday, February 3, 2021 166 Thursday, February 4, 2021 167 Date Number of the Calendar Date Friday, February 5, 2021 168 Saturday, February 6, 2021 169 Sunday, February -

Medical Drug and Step Therapy Prior Authorization List for Medicare Plus Bluesm and BCN Advantagesm Members Revised September 2021
Medical Drug and Step Therapy Prior Authorization List for Medicare Plus BlueSM and BCN AdvantageSM members Revised September 2021 This document lists the medical benefit drugs that have authorization or step therapy requirements for Medicare Advantage members. See the revision history at the end of this document for information about changes to this list. Prior authorization Submit authorization requirement effective date request through AIM HCPCS Step therapy Medicare BCN Specialty codes Generic name Trade name requirement Plus Blue Advantage NovoLogix® Health® C9076 Lisocabtagene maraleucel Breyanzi® 2/11/2021 2/11/2021 C9078 Trilaciclib Cosela™ 5/24/2021 5/24/2021 C9079 Evinacumab-dgnb Evkeeza™ 6/22/2021 6/22/2021 C9080 Melphalan flufenamide Pepaxto® 5/24/2021 5/24/2021 G2082 Esketamine Spravato® 2020 2020 (up to 56 mg plus observation) G2083 Esketamine Spravato® 2020 2020 (greater than 56 mg plus observation) 1 Medical Drug and Step Therapy Prior Authorization List for Medicare Plus BlueSM and BCN AdvantageSM members Revised September 2021 Prior authorization Submit authorization requirement effective date request through AIM HCPCS Step therapy Medicare BCN Specialty codes Generic name Trade name requirement Plus Blue Advantage NovoLogix® Health® J0129 Abatacept Orencia® 2017 2018 Try/fail Inflectra® or Renflexis®. These preferred drugs don’t require authorization. J0178 Aflibercept Eylea® 2017 2017 J0179 Brolucizumab-dbll Beovu® 2020 2020 J0180 Agalsidase beta Fabrazyme® 2017 2017 J0221 Alglucosidase alfa, 10mg Lumizyme® -

Tables-Of-Phase-3-Mabs.Pdf
Table 3. Monoclonal antibodies in Phase 2/3 or 3 clinical studies for cancer indications Most advanced Primary sponsoring company INN or code name Molecular format Target(s) phase Phase 3 indications Therapeutic area Janssen Research & Development, LLC JNJ-56022473 Humanized mAb CD123 Phase 2/3 Acute myeloid leukemia Cancer Murine IgG1, Actinium Pharmaceuticals Iomab-B radiolabeled CD45 Phase 3 Acute myeloid leukemia Cancer Humanized IgG1, Seattle Genetics Vadastuximab talirine ADC CD33 Phase 3 Acute myeloid leukemia Cancer TG Therapeutics Ublituximab Chimeric IgG1 CD20 Phase 3 Chronic lymphocytic leukemia Cancer Xencor XMAB-5574, MOR208 Humanized IgG1 CD19 Phase 2/3 Diffuse large B-cell lymphoma Cancer Moxetumomab Murine IgG1 dsFv, AstraZeneca/MedImmune LLC pasudotox immunotoxin CD22 Phase 3 Hairy cell leukemia Cancer Humanized scFv, Viventia Bio Oportuzumab monatox immunotoxin EpCAM Phase 3 Bladder cancer Cancer scFv-targeted liposome containing Merrimack Pharmaceuticals MM-302 doxorubicin HER2 Phase 2/3 Breast cancer Cancer MacroGenics Margetuximab Chimeric IgG1 HER2 Phase 3 Breast cancer Cancer Gastric cancer or gastroesophageal junction Gilead Sciences GS-5745 Humanized IgG4 MMP9 Phase 3 adenocarcinoma; ulcerative colitis (Phase 2/3) Cancer; Immune-mediated disorders Depatuxizumab Humanized IgG1, AbbVie mafodotin ADC EGFR Phase 2/3 Glioblastoma Cancer AstraZeneca/MedImmune LLC Tremelimumab Human IgG2 CTLA4 Phase 3 NSCLC, head & neck cancer, bladder cancer Cancer NSCLC, head & neck cancer, bladder cancer, breast AstraZeneca/MedImmune -

The Two Tontti Tudiul Lui Hi Ha Unit
THETWO TONTTI USTUDIUL 20170267753A1 LUI HI HA UNIT ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2017 /0267753 A1 Ehrenpreis (43 ) Pub . Date : Sep . 21 , 2017 ( 54 ) COMBINATION THERAPY FOR (52 ) U .S . CI. CO - ADMINISTRATION OF MONOCLONAL CPC .. .. CO7K 16 / 241 ( 2013 .01 ) ; A61K 39 / 3955 ANTIBODIES ( 2013 .01 ) ; A61K 31 /4706 ( 2013 .01 ) ; A61K 31 / 165 ( 2013 .01 ) ; CO7K 2317 /21 (2013 . 01 ) ; (71 ) Applicant: Eli D Ehrenpreis , Skokie , IL (US ) CO7K 2317/ 24 ( 2013. 01 ) ; A61K 2039/ 505 ( 2013 .01 ) (72 ) Inventor : Eli D Ehrenpreis, Skokie , IL (US ) (57 ) ABSTRACT Disclosed are methods for enhancing the efficacy of mono (21 ) Appl. No. : 15 /605 ,212 clonal antibody therapy , which entails co - administering a therapeutic monoclonal antibody , or a functional fragment (22 ) Filed : May 25 , 2017 thereof, and an effective amount of colchicine or hydroxy chloroquine , or a combination thereof, to a patient in need Related U . S . Application Data thereof . Also disclosed are methods of prolonging or increasing the time a monoclonal antibody remains in the (63 ) Continuation - in - part of application No . 14 / 947 , 193 , circulation of a patient, which entails co - administering a filed on Nov. 20 , 2015 . therapeutic monoclonal antibody , or a functional fragment ( 60 ) Provisional application No . 62/ 082, 682 , filed on Nov . of the monoclonal antibody , and an effective amount of 21 , 2014 . colchicine or hydroxychloroquine , or a combination thereof, to a patient in need thereof, wherein the time themonoclonal antibody remains in the circulation ( e . g . , blood serum ) of the Publication Classification patient is increased relative to the same regimen of admin (51 ) Int . -

Antibodies to Watch in 2021 Hélène Kaplona and Janice M
MABS 2021, VOL. 13, NO. 1, e1860476 (34 pages) https://doi.org/10.1080/19420862.2020.1860476 PERSPECTIVE Antibodies to watch in 2021 Hélène Kaplona and Janice M. Reichert b aInstitut De Recherches Internationales Servier, Translational Medicine Department, Suresnes, France; bThe Antibody Society, Inc., Framingham, MA, USA ABSTRACT ARTICLE HISTORY In this 12th annual installment of the Antibodies to Watch article series, we discuss key events in antibody Received 1 December 2020 therapeutics development that occurred in 2020 and forecast events that might occur in 2021. The Accepted 1 December 2020 coronavirus disease 2019 (COVID-19) pandemic posed an array of challenges and opportunities to the KEYWORDS healthcare system in 2020, and it will continue to do so in 2021. Remarkably, by late November 2020, two Antibody therapeutics; anti-SARS-CoV antibody products, bamlanivimab and the casirivimab and imdevimab cocktail, were cancer; COVID-19; Food and authorized for emergency use by the US Food and Drug Administration (FDA) and the repurposed Drug Administration; antibodies levilimab and itolizumab had been registered for emergency use as treatments for COVID-19 European Medicines Agency; in Russia and India, respectively. Despite the pandemic, 10 antibody therapeutics had been granted the immune-mediated disorders; first approval in the US or EU in 2020, as of November, and 2 more (tanezumab and margetuximab) may Sars-CoV-2 be granted approvals in December 2020.* In addition, prolgolimab and olokizumab had been granted first approvals in Russia and cetuximab saratolacan sodium was first approved in Japan. The number of approvals in 2021 may set a record, as marketing applications for 16 investigational antibody therapeutics are already undergoing regulatory review by either the FDA or the European Medicines Agency. -

Review at Launch Medication List
UnitedHealthcare® Community Plan Medical Benefit Drug List Review at Launch Medication List Last Modified Date: August 16, 2021 Table of Contents Page Instructions for Use ....................................................................... 1 Applicable Medications ................................................................. 1 List History/Revision Information ................................................. 1 Instructions for Use This Review at Launch Medication List provides the names of medications that are subject to the Medical Benefit Drug Policy titled Review at Launch for New to Market Medications and therefore, require review prior to administration. When deciding coverage, the federal, state or contractual requirements for benefit plan coverage must be referenced. The terms of the federal, state or contractual requirements for benefit plan coverage may differ greatly from the standard benefit plan upon which the aforementioned Review at Launch Drug Policy is based. In the event of a conflict, the federal, state or contractual requirements for benefit plan coverage supersede said drug policy. All reviewers must first identify member eligibility, any federal or state regulatory requirements, and the contractual requirements for benefit plan coverage prior to use. Other Policies and Coverage Determination Guidelines may apply. UnitedHealthcare reserves the right, in its sole discretion, to modify its Policies and Guidelines as necessary. Applicable Medications This medication list includes new medications that are: Food -

(12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub
US 20170172932A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub. Date: Jun. 22, 2017 (54) EARLY CANCER DETECTION AND A 6LX 39/395 (2006.01) ENHANCED IMMUNOTHERAPY A61R 4I/00 (2006.01) (52) U.S. Cl. (71) Applicant: Gholam A. Peyman, Sun City, AZ CPC .......... A61K 9/50 (2013.01); A61K 39/39558 (US) (2013.01); A61K 4I/0052 (2013.01); A61 K 48/00 (2013.01); A61K 35/17 (2013.01); A61 K (72) Inventor: sham A. Peyman, Sun City, AZ 35/15 (2013.01); A61K 2035/124 (2013.01) (21) Appl. No.: 15/143,981 (57) ABSTRACT (22) Filed: May 2, 2016 A method of therapy for a tumor or other pathology by administering a combination of thermotherapy and immu Related U.S. Application Data notherapy optionally combined with gene delivery. The combination therapy beneficially treats the tumor and pre (63) Continuation-in-part of application No. 14/976,321, vents tumor recurrence, either locally or at a different site, by filed on Dec. 21, 2015. boosting the patient’s immune response both at the time or original therapy and/or for later therapy. With respect to Publication Classification gene delivery, the inventive method may be used in cancer (51) Int. Cl. therapy, but is not limited to such use; it will be appreciated A 6LX 9/50 (2006.01) that the inventive method may be used for gene delivery in A6 IK 35/5 (2006.01) general. The controlled and precise application of thermal A6 IK 4.8/00 (2006.01) energy enhances gene transfer to any cell, whether the cell A 6LX 35/7 (2006.01) is a neoplastic cell, a pre-neoplastic cell, or a normal cell. -
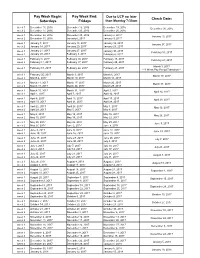
Pay Week Begin: Saturdays Pay Week End: Fridays Check Date
Pay Week Begin: Pay Week End: Due to UCP no later Check Date: Saturdays Fridays than Monday 7:30am week 1 December 10, 2016 December 16, 2016 December 19, 2016 December 30, 2016 week 2 December 17, 2016 December 23, 2016 December 26, 2016 week 1 December 24, 2016 December 30, 2016 January 2, 2017 January 13, 2017 week 2 December 31, 2016 January 6, 2017 January 9, 2017 week 1 January 7, 2017 January 13, 2017 January 16, 2017 January 27, 2017 week 2 January 14, 2017 January 20, 2017 January 23, 2017 January 21, 2017 January 27, 2017 week 1 January 30, 2017 February 10, 2017 week 2 January 28, 2017 February 3, 2017 February 6, 2017 week 1 February 4, 2017 February 10, 2017 February 13, 2017 February 24, 2017 week 2 February 11, 2017 February 17, 2017 February 20, 2017 March 3, 2017 week 1 February 18, 2017 February 24, 2017 February 27, 2017 ***1 Week Pay Period Transition*** week 1 February 25, 2017 March 3, 2017 March 6, 2017 March 17, 2017 week 2 March 4, 2017 March 10, 2017 March 13, 2017 week 1 March 11, 2017 March 17, 2017 March 20, 2017 March 31, 2017 week 2 March 18, 2017 March 24, 2017 March 27, 2017 week 1 March 25, 2017 March 31, 2017 April 3, 2017 April 14, 2017 week 2 April 1, 2017 April 7, 2017 April 10, 2017 week 1 April 8, 2017 April 14, 2017 April 17, 2017 April 28, 2017 week 2 April 15, 2017 April 21, 2017 April 24, 2017 week 1 April 22, 2017 April 28, 2017 May 1, 2017 May 12, 2017 week 2 April 29, 2017 May 5, 2017 May 8, 2017 week 1 May 6, 2017 May 12, 2017 May 15, 2017 May 26, 2017 week 2 May 13, 2017 May 19, 2017 May