Rescuing Compounds for Lesch-Nyhan Disease Identified Using Stem Cell–Based Phenotypic Screening
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mechanisms for the Regulation of Pro-Death
MECHANISMS FOR THE REGULATION OF PRO-DEATH GLYCERALDEHYDE-3-PHOSPHATE DEHYDROGENASE NUCLEAR ACCUMULATION IN RETINAL MÜLLER CELLS UNDER HIGH GLUCOSE CONDITIONS By E. CHEPCHUMBA KOECH YEGO Submitted in partial fulfillment of the requirements For the degree of Doctor of Philosophy Dissertation Advisor: Susanne Mohr, PhD Department of Physiology and Biophysics CASE WESTERN RESERVE UNIVERSITY May, 2010 2 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of ____E Chepchumba Koech Yego______________________ candidate for the _____Doctor of Philosophy (PhD)________degree *. (signed) _____Corey Smith _________ (chair of the committee) __________Cathleen Carlin__________ ______________ __________Joseph LaManna_______ ____________ __________Carole Liedtke _______________________ __________Andrea Romani_________________________ __________Michael Simonson _ _ ________ Susanne Mohr _________ (date) _____March 11th, 2010______________ *We also certify that written approval has been obtained for any proprietary material contained therein. 3 Dedication This dissertation is dedicated to my grandparents Mark Tireito *, Dinah Tireito *, Asbel Cheruiyot, Hannah Cheruiyot*, and John Korir. *Deceased 4 TABLE OF CONTENTS Dedication .......................................................................................... 3 List of Figures ..................................................................................... 8 List of Tables ................................................................................... -

Zinc Transporter Znt5/Slc30a5 Is Required for the Mast Cell–Mediated Delayed-Type Allergic Reaction but Not the Immediate-Type Reaction
ARTICLE Zinc transporter Znt5/Slc30a5 is required for the mast cell–mediated delayed-type allergic reaction but not the immediate-type reaction Keigo Nishida,1,2 Aiko Hasegawa,1,3 Susumu Nakae,4,5,6 Keisuke Oboki,4,5 Hirohisa Saito,4,5 Satoru Yamasaki,1 and Toshio Hirano1,3 1Laboratory for Cytokine Signaling, RIKEN Research Center for Allergy and Immunology, Yokohama, Kanagawa 230-0045, Japan 2Immune system, Cooperation Program, Graduate School of Frontier Biosciences, Osaka University, Osaka 565-0871, Japan 3Laboratory of Developmental Immunology and the Core Research for Evolutional Science and Technology Program (CREST) of the Japan Science and Technology Agency, Graduate School of Frontier Biosciences, Graduate School of Medicine, and WPI Immunology Frontier Research Center, Osaka University, Osaka 565-0817, Japan 4Department of Allergy and Immunology, National Research Institute for Child Health and Development, Tokyo 157-8535, Japan 5Atopy Research Center, Juntendo University, Tokyo 113-8421, Japan 6Frontier Research Initiative, Institute of Medical Science, University of Tokyo, Tokyo 108-8639, Japan Zinc (Zn) is an essential nutrient and its deficiency causes immunodeficiency. However, it remains unknown how Zn homeostasis is regulated in mast cells and if Zn transporters are involved in allergic reactions. We show that Znt5/Slc30a5 is required for contact hypersen- sitivity and mast cell–mediated delayed-type allergic response but not for immediate passive cutaneous anaphylaxis. In mast cells from Znt5/ mice, Fc receptor I (FcRI)– induced cytokine production was diminished, but degranulation was intact. Znt5 was in- volved in FcRI-induced translocation of protein kinase C (PKC) to the plasma membrane and the nuclear translocation of nuclear factor B. -

P2X and P2Y Receptors
Tocris Scientific Review Series Tocri-lu-2945 P2X and P2Y Receptors Kenneth A. Jacobson Subtypes and Structures of P2 Receptor Molecular Recognition Section, Laboratory of Bioorganic Families Chemistry, National Institute of Diabetes and Digestive and The P2 receptors for extracellular nucleotides are widely Kidney Diseases, National Institutes of Health, Bethesda, distributed in the body and participate in regulation of nearly Maryland 20892, USA. E-mail: [email protected] every physiological process.1,2 Of particular interest are nucleotide Kenneth Jacobson serves as Chief of the Laboratory of Bioorganic receptors in the immune, inflammatory, cardiovascular, muscular, Chemistry and the Molecular Recognition Section at the National and central and peripheral nervous systems. The ubiquitous Institute of Diabetes and Digestive and Kidney Diseases, National signaling properties of extracellular nucleotides acting at two Institutes of Health in Bethesda, Maryland, USA. Dr. Jacobson is distinct families of P2 receptors – fast P2X ion channels and P2Y a medicinal chemist with interests in the structure and receptors (G-protein-coupled receptors) – are now well pharmacology of G-protein-coupled receptors, in particular recognized. These extracellular nucleotides are produced in receptors for adenosine and for purine and pyrimidine response to tissue stress and cell damage and in the processes nucleotides. of neurotransmitter release and channel formation. Their concentrations can vary dramatically depending on circumstances. Thus, the state of activation of these receptors can be highly dependent on the stress conditions or disease states affecting a given organ. The P2 receptors respond to various extracellular mono- and dinucleotides (Table 1). The P2X receptors are more structurally restrictive than P2Y receptors in agonist selectivity. -

Presence of Diadenosine 5',5"'-Pl,P4-Tetraphosphate (Ap4a) In
Proc. Nati. Acad. Sci. USA Vol. 73, No. 11, pp. 3984-3988, November 1976 Biochemistry Presence of diadenosine 5',5"'-Pl,P4-tetraphosphate (Ap4A) in mammalian cells in levels varying widely with proliferative activity of the tissue: A possible positive "pleiotypic activator" (signal nucleotide/growth regulation/adenine nucleotides) ELIEZER RAPAPORT AND PAUL C. ZAMECNIK The John Collins Warren Laboratories of the Huntington Memorial Hospital of Harvard University at the Massachusetts General Hospital, Boston, Mass. 02114 Contributed by Paul C. Zamecnik, September 10, 1976 ABSTRACT An accurate assay of diadenosine 5',5"- tuting the well-known pyrophosphate exchange (Eq. 1). P',P4-tetraphosphate [A(5') pppp(5')AJ, which was shown to be formed in vitro in the backreaction of the amino acid activation pppA + aa, + Eaal :± aal-pA-Eaa1 + pp [1] step, has been developed in various cell lines in culture and in + pppA normal mouse liver or hepatoma in vivo. Use of radioactive aal-pA-Eaal labeling of acid-soluble nucleotides to high specific activity ~ 'A(5')pppp(5')A + aa, + Eaal [2] followed by chromatographic separation techniques yielded levels of Ap4A varying from 5 to 0.05 1M (from 30 pmol/mg of However, in in vitro amino acid activation systems ATP was rotein to 0.15 pmol), depending on the doubling time of the cell shown to participate in the backreaction as well (Eq. 2), yielding line or the proliferative state of the cells. The levels of Ap4A in Ap4A, which was identified as a byproduct of the first step in cells is inversely related to their doubling time, varying from protein synthesis in vitro and in vivo (Escherichia coli and rat 0.1 X 10-4 of the cellular ATP levels in slowly growing cells to liver slice) (11-15). -

Badanie Gospodarki Purynowej W Schizofrenii I Chorobach Afektywnych
1 Thomas A. Ban: Neuropsychopharmacology in Historical Perspective Collated 37 Thomas A. Ban Lithium 6. Action Janusz Rybakowski Contents Magda Malewska-Kasprzak, Agnieszka Permoda-Osip, Janusz Rybakowski: Disturbances of the purinergic system in affective disorders and schizophrenia Janusz K. Rybakowski’s additional information A commentary on Walter Felber’s paper on Lithium prevention of depression 100 years ago -- an ingenious misconception, published in 1987 Magda Malewska-Kasprzak, Agnieszka Permoda-Osip, Janusz Rybakowski: Disturbances of the purinergic system in affective disorders and schizophrenia* Abstract The purinergic system plays a role in the regulation of many psychological processes, including mood and activity. It consists of P1 receptors, with adenosine as the agonist, and P2 receptors, activated by nucleotides (e.g., adenosine 5'-triphosphate – ATP). Propounded disturbances of uric acid in affective disorders were related to the introduction of lithium for the treatment of these disorders in the 19th and 20th century. At the beginning of the 21st century, new evidence was accumulated concerning a role of uric acid in the pathogenesis and treatment of bipolar disorder (BD). In patients with BD, higher prevalence of gout and increased concentration of uric acid have been found, as well as the therapeutic activity of allopurinol, used as an adjunct to mood stabilizers, has been demonstrated in mania. 2 In recent years, the research on the role of the purinergic system in the pathogenesis and treatment of affective disorders and schizophrenia focuses on the role of adenosine (P1) receptors and nucleotide (P2) receptors. Activation of adenosine receptors is related to an antidepressant activity. Alterations of P2 receptors are also significant for the pathogenesis of affective disorders. -

Extracellular Cyclic Dinucleotides Induce Polarized Responses in Barrier Epithelial Cells by Adenosine Signaling
Extracellular cyclic dinucleotides induce polarized responses in barrier epithelial cells by adenosine signaling Denis Changa, Aaron T. Whiteleyb, Katlynn Bugda Gwilta, Wayne I. Lencera,c, John J. Mekalanosc,d,1, and Jay R. Thiagarajaha,c,1 aDivision of Gastroenterology, Hepatology and Nutrition, Boston Children’s Hospital, Harvard Medical School, Boston, MA 02115; bDepartment of Biochemistry, University of Colorado Boulder, Boulder, CO 80309; cHarvard Digestive Disease Center, Harvard Medical School, Boston, MA 02115; and dDepartment of Microbiology, Harvard Medical School, Boston, MA 02115 Contributed by John J. Mekalanos, September 14, 2020 (sent for review August 10, 2020; reviewed by Asma Nusrat and Russell E. Vance) Cyclic dinucleotides (CDNs) are secondary messengers used by pro- Although the host signaling mechanisms involved in CDN ac- karyotic and eukaryotic cells. In mammalian cells, cytosolic CDNs bind tion inside cells via activation of the STING pathway in the innate STING (stimulator of IFN gene), resulting in the production of type I immune response have been widely explored (2, 4, 7, 8), the IFN. Extracellular CDNs can enter the cytosol through several path- pathways involved in the biological activity of extracellular CDNs ways but how CDNs work from outside eukaryotic cells remains remain a new and evolving field. A number of lines of evidence poorly understood. Here, we elucidate a mechanism of action on suggest that mammalian cells release (9) or secrete (10) CDNs intestinal epithelial cells for extracellular CDNs. We found that CDNs into the extracellular environment positioning CDNs as potentially containing adenosine induced a robust CFTR-mediated chloride secre- important paracrine or autocrine signaling molecules. -
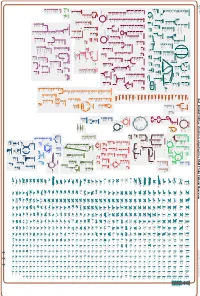
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000263195Cyc: Emticicia oligotrophica DSM 17448 Cellular Overview Connections between pathways are omitted for legibility. -

Nucleoside Tetra- and Pentaphosphates Prepared Using a Tetraphosphorylation Reagent Are Potent Inhibitors of Ribonuclease A
Nucleoside Tetra- and Pentaphosphates Prepared Using a Tetraphosphorylation Reagent Are Potent Inhibitors of Ribonuclease A The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Shepard, Scott M. et al. "Nucleoside Tetra- and Pentaphosphates Prepared Using a Tetraphosphorylation Reagent Are Potent Inhibitors of Ribonuclease A." Journal of the American Chemical Society 141, 46 (October 2019): 18400–18404 © 2019 American Chemical Society As Published http://dx.doi.org/10.1021/jacs.9b09760 Publisher American Chemical Society (ACS) Version Author's final manuscript Citable link https://hdl.handle.net/1721.1/128544 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author J Am Chem Manuscript Author Soc. Author Manuscript Author manuscript; available in PMC 2020 February 12. Published in final edited form as: J Am Chem Soc. 2019 November 20; 141(46): 18400–18404. doi:10.1021/jacs.9b09760. Nucleoside Tetra- and Pentaphosphates Prepared Using a Tetraphosphorylation Reagent Are Potent Inhibitors of Ribonuclease A Scott M. Shepard†, Ian W. Windsor†, Ronald T. Raines*, Christopher C. Cummins* Department of Chemistry, Massachusetts Institute of Technology, Cambridge Massachusetts 02139, United States Abstract Adenosine and uridine 5′-tetra- and 5′-pentaphosphates were synthesized from an activated tetrametaphosphate ([PPN]2[P4O11], [PPN]2[1], PPN = bis(triphenylphosphine)iminium) and subsequently tested for inhibition of the enzymatic activity of ribonuclease A (RNase A). Reagent [PPN]2[1] reacts with unprotected uridine and adenosine in the presence of a base under anhydrous conditions to give nucleoside tetrametaphosphates. -

P2Y6 Receptor Antagonist MRS2578 Inhibits Neutrophil Activation And
P2Y6 Receptor Antagonist MRS2578 Inhibits Neutrophil Activation and Aggregated Neutrophil Extracellular Trap Formation Induced by Gout-Associated Monosodium This information is current as Urate Crystals of September 26, 2021. Payel Sil, Craig P. Hayes, Barbara J. Reaves, Patrick Breen, Shannon Quinn, Jeremy Sokolove and Balázs Rada J Immunol published online 30 November 2016 http://www.jimmunol.org/content/early/2016/11/29/jimmun Downloaded from ol.1600766 Supplementary http://www.jimmunol.org/content/suppl/2016/11/29/jimmunol.160076 http://www.jimmunol.org/ Material 6.DCSupplemental Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 26, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published November 30, 2016, doi:10.4049/jimmunol.1600766 The Journal of Immunology P2Y6 Receptor Antagonist MRS2578 Inhibits Neutrophil Activation and Aggregated Neutrophil Extracellular Trap Formation Induced by Gout-Associated Monosodium Urate Crystals Payel Sil,* Craig P. Hayes,* Barbara J. Reaves,* Patrick Breen,† Shannon Quinn,‡ Jeremy Sokolove,x,{ and Bala´zs Rada* Human neutrophils (polymorphonuclear leukocytes [PMNs]) generate inflammatory responses within the joints of gout patients upon encountering monosodium urate (MSU) crystals. -

The Plysrs-Ap4a Pathway in Mast Cells Regulates the Switch from Host Defense to a Pathological State
International Journal of Molecular Sciences Review The pLysRS-Ap4A Pathway in Mast Cells Regulates the Switch from Host Defense to a Pathological State Sharmila Govindaraj †, Lakshmi Bhargavi Paruchuru † and Ehud Razin * Department of Biochemistry and Molecular Biology, Institute for Medical Research Israel-Canada, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem 91120, Israel; [email protected] (S.G.); [email protected] (L.B.P.) * Correspondence: [email protected] † These authors contributed equally to this work. Abstract: The innate and adaptive immune systems play an essential role in host defense against pathogens. Various signal transduction pathways monitor and balance the immune system since an imbalance may promote pathological states such as allergy, inflammation, and cancer. Mast cells have a central role in the regulation of the innate/adaptive immune system and are involved in the pathogenesis of many inflammatory and allergic diseases by releasing inflammatory mediators such as histamines, proteases, chemotactic factors, and cytokines. Although various signaling pathways are associated with mast cell activation, our discovery and characterization of the pLysRS-Ap4A signaling pathway in these cells provided an additional important step towards a full understanding of the intracellular mechanisms involved in mast cell activation. In the present review, we will discuss in depth this signaling pathway’s contribution to host defense and the pathological state. Keywords: allergic disease; mast cell activation; host defense; pathological condition; pLysRS-Ap4A Citation: Govindaraj, S.; Paruchuru, signaling L.B.; Razin, E. The pLysRS-Ap4A Pathway in Mast Cells Regulates the Switch from Host Defense to a Pathological State. -

Integrated Analysis of the Transcriptome And
International Journal of Molecular Sciences Article Integrated Analysis of the Transcriptome and Metabolome of Cecropia obtusifolia: A Plant with High Chlorogenic Acid Content Traditionally Used to Treat Diabetes Mellitus Jorge David Cadena-Zamudio 1, Pilar Nicasio-Torres 2 , Juan Luis Monribot-Villanueva 1 , José Antonio Guerrero-Analco 1 and Enrique Ibarra-Laclette 1,* 1 Instituto de Ecología, A.C. (INECOL), Red de Estudios Moleculares Avanzados (REMAV), Xalapa 91073, Veracruz, Mexico; [email protected] (J.D.C.-Z.); [email protected] (J.L.M.-V.); [email protected] (J.A.G.-A.) 2 Instituto Mexicano del Seguro Social (IMSS), Centro de Investigación Biomédica del Sur (CIBIS), Xochitepec 62790, Morelos, Mexico; [email protected] * Correspondence: [email protected]; Tel.: +52-(228)-842-1823 Received: 12 September 2020; Accepted: 9 October 2020; Published: 14 October 2020 Abstract: This investigation cultured Cecropia obtusifolia cells in suspension to evaluate the effect of nitrate deficiency on the growth and production of chlorogenic acid (CGA), a secondary metabolite with hypoglycemic and hypolipidemic activity that acts directly on type 2 diabetes mellitus. Using cell cultures in suspension, a kinetics time course was established with six time points and four total nitrate concentrations. The metabolites of interest were quantified by high-performance liquid chromatography (HPLC), and the metabolome was analyzed using directed and nondirected approaches. Finally, using RNA-seq methodology, the first transcript collection for C. obtusifolia was generated. HPLC analysis detected CGA at all sampling points, while metabolomic analysis confirmed the identity of CGA and of precursors involved in its biosynthesis. Transcriptome analysis identified differentially expressed genes and enzymes involved in the biosynthetic pathway of CGA. -

Diadenosine Oligophosphates (Apna), a Novel Class of Signalling Molecules?
View metadata,FEBS 20186 citation and similar papers at core.ac.uk FEBS Letters 427 (1998)brought to 157^163 you by CORE provided by Elsevier - Publisher Connector Minireview Diadenosine oligophosphates (ApnA), a novel class of signalling molecules? Lev L. Kisseleva;b;*, Just Justesenc, Alexey D. Wolfson1;d, Lyudmila Yu. Frolovaa;b aU248 INSERM, Institut Curie, Paris, France bEngelhardt Institute of Molecular Biology, 117984 Moscow, Russia cInstitute of Molecular and Structural Biology, University of Aarhus, Aarhus, Denmark dDepartment of Biochemistry, University of Colorado, Boulder, CO, USA Received 24 March 1998 as pleiotropically acting alarmones [5]. Further studies dem- Abstract The diadenosine oligophosphates (ApnA) were dis- covered in the mid-sixties in the course of studies on aminoacyl- onstrated a ubiquitous occurrence of ApnA in the whole spec- tRNA synthetases (aaRS). Now, more than 30 years later, about trum of organisms from bacteria to higher eukaryotes. 300 papers have been published around these substances in Although the role of ApnA in stress response remains un- attempt to decipher their role in cells. Recently, ApnA have known for eukaryotes, in bacteria ApnA binds to and inhibit emerged as intracellular and extracellular signalling molecules the oxidative stress-related proteins [6,7]. The earlier phase of implicated in the maintenance and regulation of vital cellular ApnA investigations was summarised in several reviews [8^ functions and become considered as second messengers. Great 14]. variety of physiological and pathological effects in mammalian Till the late eighties the role of ApnA in higher eukaryotes cells was found to be associated with alterations of ApnA levels remained controversial.