Quality System Regulation Overview
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anatomic Modeling Using 3D Printing: Quality Assurance and Optimization
Leng et al. 3D Printing in Medicine (2017) 3:6 DOI 10.1186/s41205-017-0014-3 RESEARCH Open Access Anatomic modeling using 3D printing: quality assurance and optimization Shuai Leng1*, Kiaran McGee1, Jonathan Morris1, Amy Alexander1, Joel Kuhlmann2, Thomas Vrieze1, Cynthia H. McCollough1 and Jane Matsumoto1 Abstract Background: The purpose of this study is to provide a framework for the development of a quality assurance (QA) program for use in medical 3D printing applications. An interdisciplinary QA team was built with expertise from all aspects of 3D printing. A systematic QA approach was established to assess the accuracy and precision of each step during the 3D printing process, including: image data acquisition, segmentation and processing, and 3D printing and cleaning. Validation of printed models was performed by qualitative inspection and quantitative measurement. The latter wasachievedbyscanningtheprintedmodelwithahighresolution CT scanner to obtain images of the printed model, which were registered to the original patient images and the distance between them was calculated on a point-by-point basis. Results: A phantom-based QA process, with two QA phantoms, was also developed. The phantoms went through the same 3D printing process as that of the patient models to generate printed QA models. Physical measurement, fit tests, and image based measurements were performed to compare the printed 3D model to the original QA phantom, with its known size and shape, providing an end-to-end assessment of errors involved in the complete 3D printing process. Measured differences between the printed model and the original QA phantom ranged from -0.32 mm to 0.13 mm for the line pair pattern. -

Construction Quality Control/Quality Assurance Plan Bozeman Landfill LFG/SVE/AI and Treatment System City of Bozeman Landfill;
Construction Quality Control/Quality Assurance Plan Bozeman Landfill LFG/SVE/AI and Treatment System City of Bozeman Landfill; Bozeman, MT #114-560487 July 10, 2015 PRESENTED TO PRESENTED BY City of Bozeman Tetra Tech, Inc. P +1-406-443-5210 PO Box 1230 303 Irene Street F +1-406-449-3729 Bozeman, MT 59711-1230 Helena, MT 59601 tetratech.com Prepared by: Mary Bell July 10, 2015 Reviewed by: Larry Cawlfield, P.E., P.H. July 10, 2015 Engineer/Hydrologist Authorized by: Larry Cawlfield, P.E., P.H. July 10, 2015 Engineer/Hydrologist Construction Quality Control/Quality Assurance Plan LFG/SVE/AI and Treatment System TABLE OF CONTENTS 1.0 INTRODUCTION ..................................................................................................................................................1 1.1 CQCQAP Organization ..................................................................................................................................1 2.0 PROJECT QA/QC ORGANIZATION ...................................................................................................................2 2.1 Responsibilities and Authorities of Key Personnel ........................................................................................2 2.1.1 Engineer of Record ...............................................................................................................................2 2.1.2 QA On-Site Supervisor .........................................................................................................................3 2.1.3 QC -

Organizational Culture and Knowledge Management Success at Project and Organizational Levels in Contracting Firms
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PolyU Institutional Repository This is the Pre-Published Version. Organizational Culture and Knowledge Management Success at Project and Organizational Levels in Contracting Firms Patrick S.W. Fong1 and Cecilia W.C. Kwok2 ABSTRACT This research focuses on contracting firms within the construction sector. It characterizes and evaluates the composition of organizational culture using four culture types (Clan, Adhocracy, Market, and Hierarchy), the strategic approach for knowledge flow, and the success of KM systems at different hierarchical levels of contracting organizations (project and parent organization level). Responses from managers of local or overseas contracting firms operating in Hong Kong were collected using a carefully constructed questionnaire survey that was distributed through electronic mail. The organizational value is analyzed in terms of the four cultural models. Clan culture is found to be the most popular at both project and organization levels, which means that the culture of contracting firms very much depends on honest communication, respect for people, trust, and cohesive relationships. On the other hand, Hierarchy 1 Associate Professor, Department of Building & Real Estate, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong (corresponding author). T: +(852) 2766 5801 F: +(852) 2764 5131 E-mail: [email protected] 2 Department of Building & Real Estate, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong. 1 culture, which focuses on stability and continuity, and analysis and control, seems to be the least favored at both levels. Another significant finding was that the two main KM strategies for knowledge flow, Codification and Personalization, were employed at both project and organization levels in equal proportion. -

Project Management © Adrienne Watt
Project Management © Adrienne Watt This work is licensed under a Creative Commons-ShareAlike 4.0 International License Original source: The Saylor Foundation http://open.bccampus.ca/find-open-textbooks/?uuid=8678fbae-6724-454c-a796-3c666 7d826be&contributor=&keyword=&subject= Contents Introduction ...................................................................................................................1 Preface ............................................................................................................................2 About the Book ..............................................................................................................3 Chapter 1 Project Management: Past and Present ....................................................5 1.1 Careers Using Project Management Skills ......................................................................5 1.2 Business Owners ...............................................................................................................5 Example: Restaurant Owner/Manager ..........................................................................6 1.2.1 Outsourcing Services ..............................................................................................7 Example: Construction Managers ..........................................................................8 1.3 Creative Services ................................................................................................................9 Example: Graphic Artists ...............................................................................................10 -
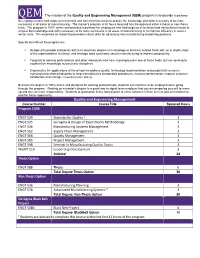
QEM the Mission of the Quality and Engineering Management
QEM The mission of the Quality and Engineering Management (QEM) program is to provide a pathway for employees who hold degrees in technical and non-technical areas to acquire the knowledge and skills necessary to be more successful in all areas of manufacturing. The master’s program is 30 hours long and has the option of either a thesis or non-thesis track. The program is 100% online and provides a pathway for employees who hold degrees in technical and nontechnical areas to acquire the knowledge and skills necessary to be more successful in all areas of manufacturing or to improve efficiency in service sector units. The emphasis on leadership provides critical skills for advancing into manufacturing leadership positions. Specific benefits of the program are: Designed to provide individuals with baccalaureate degrees in technology or business related fields with an in-depth study of the organizational, technical, and strategic tools commonly used in manufacturing to improve productivity Targeted to working professionals and other individuals who have a background in one of these fields, but are seeking to expand their knowledge across these disciplines Emphasizes the applications of these tools to address quality, technology implementation and productivity issues in manufacturing related industries to help manufacturers standardize procedures, measure performance, improve customer satisfaction and manage resources more wisely Because the degree is 100% online and designed for working professionals, students can continue to be employed while going through the program. Working on a master’s degree is a great way to signal to an employer that you are preparing yourself to move up and take on more responsibility. -

Examples of Quality Control and Quality Assurance During Construction
Examples of Quality Control and Quality Assurance During Construction Prepared by the Construction Practices Subcommittee Of APWA’s UPROW Committee Introduction The UPROW Committee requested that the Construction Practices Subcommittee research and evaluate the existing available documents related to Quality Assurance and Quality Control (QA/QC) during construction. The request initiated from the report Recommendations to Establish a New Professional / Educational / Technical Committee for Utility and Public Right-of-Way Issues, prepared by the Utility and Right-of-Way Task Force, and dated April 13, 1998. The following report documents the process that the subcommittee followed in gathering, assessing, evaluating and reporting the data and the results. Data Collection The Subcommittee chair requested that each member search for examples of specifications, guidelines, manuals, programs or other written information, which outlined methods or requirements of QA/QC during construction. The members of the subcommittee were located in various regions across the entire country, from governmental (local, state and federal), industrial, commercial and private consulting backgrounds. A total of nine documents were received and reviewed. While the number of documents was not expansive, it was the opinion of the subcommittee that the sources of these documents have extensive background in construction quality control and therefore these documents cover the topic adequately. It is likely that other agencies have very good examples of standards but the subcommittee was unable to obtain other documents in their search. Analysis and Evaluation Prior to review the documents, members of the subcommittee developed a list of criteria to allow a subjective evaluation of the sample documents. These criteria included the following factors: · Quality Control Organization Items considered: The classifications of the employees included within the structure of the organization. -

Designing, Developing and Implementing a Management System: an Overview (April 2010)
Designing, Developing and Implementing a Management System: An Overview (April 2010) Host: Emily Smorol, Corporate Environmental Affairs Speakers: Patrick Aurrichio, Program Manager, Corporate Environmental Affairs Emily: Welcome to our IBM Podcast titled, “Designing, Developing, and Implementing a Management System: An Overview.” My name is Emily Smorol and I work in IBM’s Corporate Environmental Affairs and Product Safety organization. There is an accompanying presentation available to download for this Podcast. For those following along with the presentation slides, please go to slide 2. The purpose of this Podcast is to discuss what a management system is and to provide an overview of its basic components. This Podcast is not meant to be all-inclusive but rather provide an approach to designing, developing, and implementing a management system in a sustained manner that has been successful for us – and which also may prove to be helpful to others setting out to do similarly. Today, we are going to be speaking with IBM’s Corporate Environmental Affairs Program Manager, Patrick Aurrichio. Patrick was instrumental in developing and implementing IBM’s global Environmental Management System. Recognizing that our audience comes from a diverse background – those with experience or familiarity with management systems and those for whom this may be a new concept – Patrick, how would you describe what a management system is? Patrick: Thank you Emily – and welcome to those joining the podcast. If you’re following along with the slide presentation, please turn to slide 3. First I’d like to mention and want you to understand that there is nothing overwhelmingly special or difficult with designing or developing a management system. -

Project Information Management PROJECT MANAGEMENT for DEVELOPMENT ORGANIZATIONS Project Information Management
pm4dev, 2016 –management for development series © Project Information Management PROJECT MANAGEMENT FOR DEVELOPMENT ORGANIZATIONS Project Information Management PROJECT MANAGEMENT FOR DEVELOPMENT ORGANIZATIONS A methodology to manage development projects for international humanitarian assistance and relief organizations © PM4DEV 2016 Our eBook is provided free of charge on the condition that it is not copied, modified, published, sold, re-branded, hired out or otherwise distributed for commercial purposes. Please give appropriate citation credit to the authors and to PM4DEV. Feel free to distribute this eBook to any one you like, including peers, managers, and organizations to assist in their project management activities. www.pm4dev.com Project Information Management INTRODUCTION “If you fail to plan, you plan to fail.” “.. A major weakness is the ability of project staff to utilize their logframe for designing a coherent and integrated, overall information system, where a manageable and limited number of feasible information activities are planned, which together will ensure that effective effect and impact level monitoring will occur. It is typical for projects to end up collecting too much rather than too little information. Frequently though, much of this information is not relevant to monitoring the results and impacts for which the project is accountable, and that which is, is not collected sufficiently reliably or regularly. By restricting the number, but improving the quality and reliability of their major information gathering activities, projects will much improve their information systems.” CARE International EDIAIS Case Study Project Information Management Plan Detailed planning is critical to the development of usable, high quality information deliverables that meet the needs of internal and external information users. -

Leadership and Programme Management in Infection Prevention and Control: a Trainer’S Guide
Advanced Infection Prevention and Control Training Issue version 2018 Leadership and programme management in infection prevention and control: a trainer’s guide Outline of the module The Leadership and programme management in infection prevention and control (IPC) advanced training module is part of a package designed for advanced IPC focal persons working in low-resource settings. It is designed to support the implementation of the World Health Organization (WHO) Guidelines on core components of IPC programmes at the national and acute health care facility level1 as part of a multifaceted approach to capacity building. Target audience This training is designed for individuals and teams who are intending to occupy a senior leadership position in IPC at the national, sub-national and health facility level. Trainees are expected to possess at least basic experience and competence in IPC and could include (not exhaustive) IPC professionals, IPC hospital teams, facility administrators, hospital epidemiologists, microbiologists or other relevant health care professionals. The advanced training package complements a basic training package intended for all frontline healthcare workers. Objectives of the module The objectives of the module are to equip the advanced IPC focal person to: 1. define the roles and responsibilities of an IPC focal person; 2. describe the requirements of an IPC programme according to the WHO core components’ guidelines; 3. demonstrate key leadership skills; 4. demonstrate conflict management and communication skills; 5. advocate for IPC as a priority in health care, as well as describing the need for synergies with other programmes; 6. foster teamwork; 7. lead project development, management and budget planning necessary for an IPC programme; 8. -

Defining the Quality of Business Processes
Defining the Quality of Business Processes Robert Heinrich1 and Barbara Paech2 Abstract: Business process models are used to gain a joint understanding of complex processes. Often they are applied in change projects where either the supporting IT or the processes themselves or both are to be improved. So it is an important question to assess the quality of the modeled business processes. However, so far there is no standard definition of the quality of a business process. Furthermore, business process models are not tuned to capture quality aspects. The goal of our work is to collect important quality characteristics and attributes of processes and to enhance business process modeling languages with means to express these attributes. This paper is a first step in this direction. We define a first set of quality characteristics, attributes and measures for these attributes in a business process. As an example we evaluate how well these measures can be expressed in a BPMN model. 1Introduction Business process models aim at providing a joint understanding of business processes. Therefore, they typically cover information about structure and behavior like description of activities or decisions within the process. But they do not aim at providing quality information. Quality information, for example, is information about the reliability or the usability of a business process. That information is of high interest for organizations because business process models are often used in change projects where either the supporting IT or the processes themselves or both are to be enhanced. Furthermore, the quality of a business process highly affects the success of an organization. -

LFS – Logistics Focused Solutions Integrated Total Solutions
LFS – Logistics Focused Solutions Integrated total solutions EPS – Ehrhardt + Partner Solutions DWC-LLC | Software Systems for Warehouse Logistics P.O.Box 644 300 | Dubai, UAE | Phone (+971) 4-870 1000 | Fax (+971) 4-870 1050 [email protected] | www.ehrhardt-partner.ae Warehouse-Management by E+P The internationally leading expert for warehouse logistics Ehrhardt + Partner GmbH & Co. KG | Software Systems for Warehouse Logistics Alte Römerstraße 3 | D-56154 Boppard-Buchholz | Germany | Phone (+49) 67 42-87 27 0 | Fax (+49) 67 42-87 27 50 [email protected] | www.ehrhardt-partner.com Contents LFS – Logistics Focused Solutions ................................................................................... 3 A system for all requirements The warehouse management system for all platforms Open system solution for your IT architecture LFS as a central solution for your warehouse logistics .................................................... 6 The total solution for your individual task definitions Central software installation for different countries and warehouses LFS as a hosting and cloud solution Setting standards with innovations ................................................................................... 8 Pick-by-Voice Voice service and support RFID Multi-order picking LFS – a system solution all types of work ...................................................................... 10 Professional connection of automatic warehouse units Systematic warehouse modernisation Sound advice and scheduled implementation -

How Is Your Leadership Changing? Margaret Wheatley ©2005
How is Your Leadership Changing? Margaret Wheatley ©2005 I'm sad to report that in the past few years, ever since uncertainty became our insistent 21st century companion, leadership has taken a great leap backwards to the familiar territory of command and control. Some of this was to be expected, because humans usually default to the known when confronted with the unknown. Some of it was a surprise, because so many organizations had focused on innovation, quality, learning organizations, and human motivation. How did they fail to learn that whenever you impose control on people and situations, you only succeed in turning people into non-creative, shut-down and cynical workers? The destructive impact of command and control The dominance of command and control is having devastating impacts. There has been a dramatic increase in worker disengagement, few organizations are succeeding at solving problems, and leaders are being scapegoated and fired. Most people associate command and control leadership with the military. Years ago, I worked for the U.S. Army Chief of Staff. I, like most people, thought I'd see command and control leadership there. The great irony is that the military learned long ago that, if you want to win, you have to engage the intelligence of everyone involved in the battle. The Army had a visual reminder of this when, years ago, they developed new tanks and armored vehicles that traveled at unprecedented speeds of fifty miles an hour. When first used in battle during the first Gulf War, several times troops took off on their own, speeding across the desert at high speed.