Identifying Mirna Sponge Modules Using Biclustering and Regulatory Scores Junpeng Zhang1*†, Thuc D Le2†, Lin Liu2 and Jiuyong Li2*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The KMT1A-GATA3-STAT3 Circuit Is a Novel Self-Renewal Signaling of Human Bladder Cancer Stem Cells Zhao Yang1, Luyun He2,3, Kais
The KMT1A-GATA3-STAT3 circuit is a novel self-renewal signaling of human bladder cancer stem cells Zhao Yang1, Luyun He2,3, Kaisu Lin4, Yun Zhang1, Aihua Deng1, Yong Liang1, Chong Li2, 5, & Tingyi Wen1, 6, 1CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China 2Core Facility for Protein Research, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China 3CAS Key Laboratory of Infection and Immunity, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China 4Department of Oncology, the Second Affiliated Hospital of Soochow University, Suzhou 215000, China 5Beijing Jianlan Institute of Medicine, Beijing 100190, China 6Savaid Medical School, University of Chinese Academy of Sciences, Beijing 100049, China Correspondence author: Tingyi Wen, e-mail: [email protected] Chong Li, e-mail: [email protected] Supplementary Figure S1. Isolation of human bladder cancer stem cells. BCMab1 and CD44 were used to isolate bladder cancer stem cells (BCSCs: BCMab1+CD44+) and bladder cancer non-stem cells (BCNSCs: BCMab1-CD44-) from EJ, samples #1 and #2 by flow cytometry. Supplementary Figure S2. Gene ontology analysis of downregulated genes of human BCSCs. (A) Pathway enrichment of 103 downregulated genes in BCSCs. (B) The seven downregulated genes in BCSCs participating in centromeric heterochromatin, mRNA-3’-UTR binding and translation regulator activity signaling pathways were validated by qRT-PCR. Data are presented as mean ± SD. P < 0.05; P < 0.01. Supplementary Figure S3. The expression of KMT1A is higher in human BC than that in peri-tumor tissues. (A) The expression of KMT1A was higher in BC samples than that in peri-tumors as assessed by immunohistochemistry, Scale bar = 50 m. -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

WNT16 Is a New Marker of Senescence
Table S1. A. Complete list of 177 genes overexpressed in replicative senescence Value Gene Description UniGene RefSeq 2.440 WNT16 wingless-type MMTV integration site family, member 16 (WNT16), transcript variant 2, mRNA. Hs.272375 NM_016087 2.355 MMP10 matrix metallopeptidase 10 (stromelysin 2) (MMP10), mRNA. Hs.2258 NM_002425 2.344 MMP3 matrix metallopeptidase 3 (stromelysin 1, progelatinase) (MMP3), mRNA. Hs.375129 NM_002422 2.300 HIST1H2AC Histone cluster 1, H2ac Hs.484950 2.134 CLDN1 claudin 1 (CLDN1), mRNA. Hs.439060 NM_021101 2.119 TSPAN13 tetraspanin 13 (TSPAN13), mRNA. Hs.364544 NM_014399 2.112 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 2.070 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 2.026 DCBLD2 discoidin, CUB and LCCL domain containing 2 (DCBLD2), mRNA. Hs.203691 NM_080927 2.007 SERPINB2 serpin peptidase inhibitor, clade B (ovalbumin), member 2 (SERPINB2), mRNA. Hs.594481 NM_002575 2.004 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.989 OBFC2A Oligonucleotide/oligosaccharide-binding fold containing 2A Hs.591610 1.962 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.947 PLCB4 phospholipase C, beta 4 (PLCB4), transcript variant 2, mRNA. Hs.472101 NM_182797 1.934 PLCB4 phospholipase C, beta 4 (PLCB4), transcript variant 1, mRNA. Hs.472101 NM_000933 1.933 KRTAP1-5 keratin associated protein 1-5 (KRTAP1-5), mRNA. Hs.534499 NM_031957 1.894 HIST2H2BE histone cluster 2, H2be (HIST2H2BE), mRNA. Hs.2178 NM_003528 1.884 CYTL1 cytokine-like 1 (CYTL1), mRNA. Hs.13872 NM_018659 tumor necrosis factor receptor superfamily, member 10d, decoy with truncated death domain (TNFRSF10D), 1.848 TNFRSF10D Hs.213467 NM_003840 mRNA. -

The Role of Natural Selection in Human Evolution – Insights from Latin America
Genetics and Molecular Biology, 39, 3, 302-311 (2016) Copyright © 2016, Sociedade Brasileira de Genética. Printed in Brazil DOI: http://dx.doi.org/10.1590/1678-4685-GMB-2016-0020 Review Article The role of natural selection in human evolution – insights from Latin America Francisco M. Salzano1 1Departamento de Genética, Instituto de Biociências, Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brazil. Abstract A brief introduction considering Darwin’s work, the evolutionary synthesis, and the scientific biological field around the 1970s and subsequently, with the molecular revolution, was followed by selected examples of recent investiga- tions dealing with the selection-drift controversy. The studies surveyed included the comparison between essential genes in humans and mice, selection in Africa and Europe, and the possible reasons why females in humans remain healthy and productive after menopause, in contrast with what happens in the great apes. At the end, selected exam- ples of investigations performed in Latin America, related to the action of selection for muscle performance, acetylation of xenobiotics, high altitude and tropical forest adaptations were considered. Despite dissenting views, the influence of positive selection in a considerable portion of the human genome cannot presently be dismissed. Keywords: natural selection, human evolution, population genetics, human adaptation, history of genetics. Received: February 10, 2016; Accepted: May 8, 2016. History deniably indicated that we had derived from -

As a Risk Gene for Schizophrenia X Chen Et Al
Molecular Psychiatry (2011) 16, 1117–1129 & 2011 Macmillan Publishers Limited All rights reserved 1359-4184/11 www.nature.com/mp ORIGINAL ARTICLE GWA study data mining and independent replication identify cardiomyopathy-associated 5 (CMYA5)asa risk gene for schizophrenia X Chen1,2, G Lee1, BS Maher1, AH Fanous1,3,4, J Chen1, Z Zhao5, A Guo5, E van den Oord1,6, PF Sullivan7, J Shi8, DF Levinson8, PV Gejman9, A Sanders9, J Duan9, MJ Owen10, NJ Craddock10, MC O’Donovan10, J Blackman10, D Lewis10, GK Kirov10, W Qin11, S Schwab11, D Wildenauer11, K Chowdari12, V Nimgaonkar12, RE Straub13, DR Weinberger13, FA O’Neill14, D Walsh15, M Bronstein16, A Darvasi16, T Lencz17, AK Malhotra17, D Rujescu18, I Giegling18, T Werge19, T Hansen19, A Ingason19, MM No¨ethen20, M Rietschel21,22, S Cichon20,23, S Djurovic24, OA Andreassen24, RM Cantor25, R Ophoff 25,26, A Corvin27, DW Morris27, M Gill27, CN Pato28, MT Pato28, A Macedo29, HMD Gurling30, A McQuillin30, J Pimm30, C Hultman31, P Lichtenstein31, P Sklar32,33, SM Purcell32,33, E Scolnick32,33, D St Clair 34, DHR Blackwood35 and KS Kendler1,2 and the GROUP investigators, the International Schizophrenia Consortium36 1Department of Psychiatry, Virginia Institute for Psychiatric and Behavioral Genetics, Virginia Commonwealth University, Richmond, VA, USA; 2Department of Molecular and Human Genetics, Virginia Commonwealth University, Richmond, VA, USA; 3Washington VA Medical Center, Washington, DC, USA; 4Department of Psychiatry, Georgetown University School of Medicine, Washington, DC, USA; 5Departments -

Sequencing, Analysis, and Annotation of Expressed Sequence Tags for Camelus Dromedarius
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Faculty Publications from the Department of Electrical & Computer Engineering, Department Electrical and Computer Engineering of 2009 Sequencing, Analysis, and Annotation of Expressed Sequence Tags for Camelus dromedarius Abdulaziz M. Al-Swailem King Abdulaziz City for Science and Technology Maher M. Shehata King Abdulaziz City for Science and Technology Faisel M. Abu-Duhier King Abdulaziz City for Science and Technology Essam J. Al-Yamani King Abdulaziz City for Science and Technology Khalid A. Al-Busadah King Faisal University See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/electricalengineeringfacpub Part of the Computer Engineering Commons, and the Electrical and Computer Engineering Commons Al-Swailem, Abdulaziz M.; Shehata, Maher M.; Abu-Duhier, Faisel M.; Al-Yamani, Essam J.; Al-Busadah, Khalid A.; Al-Arawi, Mohammed S.; Al-Khider, Ali Y.; Al-Muhaimeed, Abdullah N.; Al-Qahtani, Fahad H.; Manee, Manee M.; Al-Shomrani, Badr M.; Al-Qhtani, Saad M.; Al-Harthi, Amer S.; Akdemir, Kadir C.; Inan, Mehmet S.; and Otu, Hasan H., "Sequencing, Analysis, and Annotation of Expressed Sequence Tags for Camelus dromedarius" (2009). Faculty Publications from the Department of Electrical and Computer Engineering. 426. https://digitalcommons.unl.edu/electricalengineeringfacpub/426 This Article is brought to you for free and open access by the Electrical & Computer Engineering, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Faculty Publications from the Department of Electrical and Computer Engineering by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Abdulaziz M. Al-Swailem, Maher M. -

HHS Public Access Author Manuscript
HHS Public Access Author manuscript Author Manuscript Author ManuscriptOncogene Author Manuscript. Author manuscript; Author Manuscript available in PMC 2015 July 02. Published in final edited form as: Oncogene. 2015 January 2; 34(1): 94–103. doi:10.1038/onc.2013.523. Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma Paul N. Valdmanis1,2, Biswajoy Roy-Chaudhuri1,2, Hak Kyun Kim1,2, Leanne C. Sayles3, Yanyan Zheng3, Chen-Hua Chuang2,4,5, Deborah R. Caswell2,4,5, Kirk Chu1,2, Monte M. Winslow2,4,5, E. Alejandro Sweet-Cordero1,3,5, and Mark A. Kay1,2 1Department of Pediatrics, Stanford University, Stanford, CA, USA 2Department of Genetics, Stanford University, Stanford, CA, USA 3Division of Hematology/Oncology, Department of Pediatrics, Stanford University, Stanford, CA, USA 4Cancer Biology Program, Stanford University, Stanford, CA, USA 5Stanford Cancer Institute, Stanford University, Stanford, CA, USA Abstract Mice in which lung epithelial cells can be induced to express an oncogenic KrasG12D develop lung adenocarcinomas in a manner analogous to humans. A myriad of genetic changes accompany lung adenocarcinomas, many of which are poorly understood. To get a comprehensive understanding of both the transcriptional and post-transcriptional changes that accompany lung adenocarcinomas, we took an omics approach in profiling both the coding genes and the non- coding small RNAs in an induced mouse model of lung adenocarcinoma. RNAseq transcriptome analysis of KrasG12D tumors from F1 hybrid mice revealed features specific to tumor samples. This includes the repression of a network of GTPase related genes (Prkg1, Gnao1 and Rgs9) in tumor samples and an enrichment of Apobec1-mediated cytosine to uridine RNA editing. -

Sleeping Beauty Transposon Mutagenesis As a Tool for Gene Discovery in the NOD Mouse Model of Type 1 Diabetes
MUTANT SCREEN REPORT Sleeping Beauty Transposon Mutagenesis as a Tool for Gene Discovery in the NOD Mouse Model of Type 1 Diabetes Colleen M. Elso,*,1 Edward P. F. Chu,*,†,1 May A. Alsayb,*,†,1 Leanne Mackin,* Sean T. Ivory,* Michelle P. Ashton,*,†,2 Stefan Bröer,‡ Pablo A. Silveira,§ and Thomas C. Brodnicki*,†,3 *Immunology and Diabetes Unit, St. Vincent’s Institute, Fitzroy, Victoria 3065, Australia, †Department of Medicine, University of Melbourne, Parkville, Victoria 3010, Australia, ‡Research School of Biology, The Australian National University, Canberra, § ACT 2601, Australia, and Dendritic Cell Research, ANZAC Research Institute, Concord, New South Wales 2139, Australia ABSTRACT A number of different strategies have been used to identify genes for which genetic variation KEYWORDS contributes to type 1 diabetes (T1D) pathogenesis. Genetic studies in humans have identified .40 loci that affect forward genetics the risk for developing T1D, but the underlying causative alleles are often difficult to pinpoint or have subtle reverse genetics biological effects. A complementary strategy to identifying “natural” alleles in the human population is to engi- Slc16a10 neer “artificial” alleles within inbred mouse strains and determine their effect on T1D incidence. We describe the Serinc1 use of the Sleeping Beauty (SB) transposon mutagenesis system in the nonobese diabetic (NOD) mouse strain, amino acid which harbors a genetic background predisposed to developing T1D. Mutagenesis in this system is random, but a transporter green fluorescent protein (GFP)-polyA gene trap within the SB transposon enables early detection of mice harboring transposon-disrupted genes. The SB transposon also acts as a molecular tag to, without additional breeding, efficiently identify mutated genes and prioritize mutant mice for further characterization. -
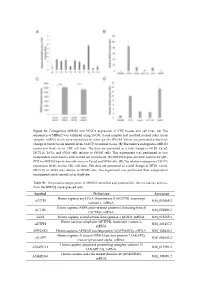
Figure S1. Endogenous MIR45
Figure S1. Endogenous MIR452 and VEGFA expression in CRC tissues and cell lines. (A) The expression of MIR452 was validated using 10 CRC tissue samples and matched normal colon tissue samples. miRNA levels were normalized to colon-specific RNU48. Values are presented as the fold- change in tumor tissue relative levels (ΔΔCT) to normal tissue. (B) The relative endogenous MIR452 expression levels in six CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, LoVo, and SW48 cells relative to SW480 cells. This experiment was performed as two independent experiments, each carried out in triplicate. (C) MIR452 expression level analysis by qRT- PCR for MIR452 transfection efficiency in Caco2 and SW48 cells. (D) The relative endogenous VEGFA expression levels in five CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, or SW48 cells relative to SW480 cells. This experiment was performed three independent experiments, each carried out in duplicate. Table S1. The putative target genes of MIR452 identified and predicted by the microarray analysis from the MIR452 overexpressed cells. Symbol Definition Accession Homo sapiens acyl-CoA thioesterase 8 (ACOT8), transcript ACOT8 NM_005469.2 variant 1, mRNA. Homo sapiens ARP6 actin-related protein 6 homolog (yeast) ACTR6 NM_022496.3 (ACTR6), mRNA. ADI1 Homo sapiens acireductone dioxygenase 1 (ADI1), mRNA. NM_018269.1 Homo sapiens aftiphilin (AFTPH), transcript variant 1, AFTPH NM_203437.2 mRNA. AHNAK2 Homo sapiens AHNAK nucleoprotein 2 (AHNAK2), mRNA. NM_138420.2 Homo sapiens A kinase (PRKA) anchor protein 7 (AKAP7), AKAP7 NM_004842.2 transcript variant alpha, mRNA. Homo sapiens anaphase promoting complex subunit 13 ANAPC13 NM_015391.2 (ANAPC13), mRNA. -

Improved De Novo Genome Assembly: Linked-Read Sequencing Combined with Optical Mapping Produce a High Quality Mammalian Genome at Relatively Low Cost
bioRxiv preprint doi: https://doi.org/10.1101/128348; this version posted April 19, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Improved de novo Genome Assembly: Linked-Read Sequencing Combined with Optical Mapping Produce a High Quality Mammalian Genome at Relatively Low Cost Mohr DW1*, Naguib A2, Weisenfeld NI3, Kumar V3, Shah P3, Church DM3, Jaffe D3 and AF Scott1* 1Genetic Resources Core Facility, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21287 2BioNano Genomics, Inc., 9640 ToWne Centre Dr, Suite 100, San Diego, CA 92121 310X Genomics, Inc., 7068 Koll Center ParkWay #401, Pleasanton, CA 94566 Abstract: Current short-read methods have come to dominate genome sequencing because they are cost-effective, rapid, and accurate. HoWever, short reads are most applicable when data can be aligned to a known reference. Two new methods for de novo assembly are linked-reads and restriction-site labeled optical maps. We combined commercial applications of these technologies for genome assembly of an endangered mammal, the HaWaiian Monk seal. We show that the linked-reads produced With 10X Genomics Chromium chemistry and assembled With Supernova v1.1 software produced scaffolds with an N50 of 22.23 Mbp with the longest individual scaffold of 84.06 Mbp. When combined With Bionano Genomics optical maps using Bionano RefAligner, the scaffold N50 increased to 29.65 Mbp for a total of 170 hybrid scaffolds, the longest of which was 84.78 Mbp. -

Upregulation of the Microrna Cluster at the Dlk1-Dio3 Locus in Lung Adenocarcinoma
Oncogene (2015) 34, 94–103 & 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc ORIGINAL ARTICLE Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma PN Valdmanis1,2, B Roy-Chaudhuri1,2, HK Kim1,2, LC Sayles3, Y Zheng3, C-H Chuang2,4,5, DR Caswell2,4,5, K Chu1,2, Y Zhang1,2, MM Winslow2,4,5, EA Sweet-Cordero1,3,5 and MA Kay1,2 Mice in which lung epithelial cells can be induced to express an oncogenic KrasG12D develop lung adenocarcinomas in a manner analogous to humans. A myriad of genetic changes accompany lung adenocarcinomas, many of which are poorly understood. To get a comprehensive understanding of both the transcriptional and post-transcriptional changes that accompany lung adenocarcinomas, we took an omics approach in profiling both the coding genes and the non-coding small RNAs in an induced mouse model of lung adenocarcinoma. RNAseq transcriptome analysis of KrasG12D tumors from F1 hybrid mice revealed features specific to tumor samples. This includes the repression of a network of GTPase-related genes (Prkg1, Gnao1 and Rgs9) in tumor samples and an enrichment of Apobec1-mediated cytosine to uridine RNA editing. Furthermore, analysis of known single- nucleotide polymorphisms revealed not only a change in expression of Cd22 but also that its expression became allele specific in tumors. The most salient finding, however, came from small RNA sequencing of the tumor samples, which revealed that a cluster of B53 microRNAs and mRNAs at the Dlk1-Dio3 locus on mouse chromosome 12qF1 was markedly and consistently increased in tumors.