Directed Evolution of Enzymes Using Ultrahigh-Throughput Screening
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Supramolecular-Hydrogel-Encapsulated Hemin As an Artificial Enzyme to Mimic Peroxidase This Work Was Partially Supported by R
Angewandte Chemie DOI: 10.1002/anie.200700404 Enzyme Mimetics A Supramolecular-Hydrogel-Encapsulated Hemin as an Artificial Enzyme to Mimic Peroxidase** Qigang Wang, Zhimou Yang, Xieqiu Zhang, Xudong Xiao, Chi K. Chang,* and Bing Xu* A challenge in chemistry is an artificial enzyme[1] that mimics biomineralization,[15] and as biomaterials for wound heal- the functions of the natural system but is simpler than ing.[13] The application of supramolecular hydrogels as the proteins. The intensive development of artificial enzymes that skeletons of artificial enzymes has yet to be explored. Similar use a variety of matrices,[2] the rapid progress in supramolec- to peptide chains that form active sites in enzymes, the self- ular gels,[3] and the apparent “superactivity” exhibited by assembled nanofibers of amino acids in the supramolecular supramolecular hydrogel-immobilized enzymes,[4] prompted hydrogels could act as the matrices of artificial enzymes. Thus, us to evaluate whether supramolecular hydrogels will the supramolecular-hydrogel systems serve two functions: improve the activity of artificial enzymes for catalyzing 1) as the skeletons of the artificial enzyme to aid the function reactions in water or in organic media. To demonstrate the of the active site (e.g., hemin) and, 2) as the immobilization concept, we used hemin as the prosthetic group to mimic carriers to facilitate the recovery of the catalysts in practical peroxidase, a ubiquitous enzyme that catalyzes the oxidation applications. of a broad range of organic and inorganic substrates by Herein, we mixed hemin chloride (3) into the hydrogel hydrogen peroxide or organic peroxides. The structures of the formed by the self-assembly of two simple derivatives of active site as well as the reaction mechanism of peroxidases amino acids (1 and 2). -

Philosophy of Science and Philosophy of Chemistry
Philosophy of Science and Philosophy of Chemistry Jaap van Brakel Abstract: In this paper I assess the relation between philosophy of chemistry and (general) philosophy of science, focusing on those themes in the philoso- phy of chemistry that may bring about major revisions or extensions of cur- rent philosophy of science. Three themes can claim to make a unique contri- bution to philosophy of science: first, the variety of materials in the (natural and artificial) world; second, extending the world by making new stuff; and, third, specific features of the relations between chemistry and physics. Keywords : philosophy of science, philosophy of chemistry, interdiscourse relations, making stuff, variety of substances . 1. Introduction Chemistry is unique and distinguishes itself from all other sciences, with respect to three broad issues: • A (variety of) stuff perspective, requiring conceptual analysis of the notion of stuff or material (Sections 4 and 5). • A making stuff perspective: the transformation of stuff by chemical reaction or phase transition (Section 6). • The pivotal role of the relations between chemistry and physics in connection with the question how everything fits together (Section 7). All themes in the philosophy of chemistry can be classified in one of these three clusters or make contributions to general philosophy of science that, as yet , are not particularly different from similar contributions from other sci- ences (Section 3). I do not exclude the possibility of there being more than three clusters of philosophical issues unique to philosophy of chemistry, but I am not aware of any as yet. Moreover, highlighting the issues discussed in Sections 5-7 does not mean that issues reviewed in Section 3 are less im- portant in revising the philosophy of science. -

Fe(III) Porphyrin Metal–Organic Framework As an Artificial Enzyme Mimics and Its Application in Biosensing of Glucose and H2O2
Source: Aghayan, M., Mahmoudi, A., Nazari, K., Dehghanpour, S., Sohrabi, S., Sazegar, M.R. and Mohammadian- Tabrizi, N., 2019. Fe (III) porphyrin metal–organic framework as an artificial enzyme mimics and its application in biosensing of glucose and H2O2. Journal of Porous Materials, 26(5), pp.1507-1521. DOI: 10.1007/s10934-019-00748-4 Fe(III) porphyrin metal–organic framework as an artificial enzyme mimics and its application in biosensing of glucose and H2O2 Morvarid Aghayan1, Ali Mahmoudi1, Khodadad Nazari2, Saeed Dehghanpour3, Samaneh Sohrabi3, Mohammad Reza Sazegar1, Navid Mohammadian‑Tabrizi4 1 Department of Chemistry, Faculty of Science, North Tehran Branch, Islamic Azad University, Tehran, Iran 2 Research Institute of Petroleum Industry, N.I.O.C, Tehran, Iran 3 Department of Chemistry, Faculty of Science, Alzahra University, Tehran, Iran 4 Department of Chemistry, Faculty of Science, University of Tehran, Tehran, Iran Abstract Metal–organic frameworks with diverse structures and unique properties have demonstrated that can be an ideal substitute for natural enzymes in colorimetric sensing platform for analyte detection in various fields such as environmental chemistry, biotechnology and clinical diagnostics, which have attracted the scientist’s attention, recently. In this study, a porous coordination network (denoted as PCN-222) was synthesized as a new biomimetic material from an iron linked tetrakis (4-carboxyphenyl) porphyrin (named as Fe-TCPP) as a heme-like ligand and Zr6 linker as a node. This catalyst shows the peroxidase and catalase activities clearly. The mechanism of peroxidase activity for PCN-222 was investigated using the spectrophotometric methods and its activity was compared with the other nanoparticles which, the results showed a higher activity than the other catalysts. -

A Mechanistic Rationale Approach Revealed the Unexpected
A Mechanistic Rationale Approach Revealed the Unexpected Chemoselectivity of an Artificial Ru-Dependent Oxidase: A Dual Experimental/Theoretical Approach Sarah Lopez, David Michael Mayes, Serge Crouzy, Christine Cavazza, Chloé Leprêtre, Yohann Moreau, Nicolai Burzlaff, Caroline Marchi-Delapierre, Stéphane Ménage To cite this version: Sarah Lopez, David Michael Mayes, Serge Crouzy, Christine Cavazza, Chloé Leprêtre, et al.. A Mech- anistic Rationale Approach Revealed the Unexpected Chemoselectivity of an Artificial Ru-Dependent Oxidase: A Dual Experimental/Theoretical Approach. ACS Catalysis, American Chemical Society, 2020, 10 (10), pp.5631-5645. 10.1021/acscatal.9b04904. hal-02865406 HAL Id: hal-02865406 https://hal.archives-ouvertes.fr/hal-02865406 Submitted on 26 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. A Mechanistic Rationale Approach Revealed the Unexpected Chemoselectivity of an Artificial Ru-dependent Oxidase - A Dual Experimental/Theoretical Approach By Sarah Lopez,1,2 David Michael Mayes,1 Serge Crouzy,1 Christine Cavazza,1 Chloé Leprêtre,1 Yohann Moreau,1 Nicolai Burzlaff,3 Caroline Marchi-Delapierre*1 and Stéphane Ménage1 [1] Univ. Grenoble Alpes, CEA, CNRS, IRIG, CBM, F-38000 Grenoble, France. [2] Univ. Grenoble-Alpes, DCM-SeRCO, Grenoble, France. -

Catalytic, Theoretical, and Biological Investigation of an Enzyme Mimic Model
Turkish Journal of Chemistry Turk J Chem (2021) 45: 1270-1278 http://journals.tubitak.gov.tr/chem/ © TÜBİTAK Research Article doi:10.3906/kim-2104-51 Catalytic, theoretical, and biological investigation of an enzyme mimic model Gülcihan GÜLSEREN* Department of Molecular Biology and Genetics, Faculty of Agriculture and Natural Sciences, Konya Food and Agriculture University, Turkey Received: 20.04.2021 Accepted/Published Online: 12.06.2021 Final Version: 27.08.2021 Abstract: Artificial catalyst studies were always stayed at the kinetics investigation level, in this work bioactivity of designed catalyst were shown by the induction of biomineralization of the cells, indicating the possible use of enzyme mimics for biological applications. The development of artificial enzymes is a continuous quest for the development of tailored catalysts with improved activity and stability. Understanding the catalytic mechanism is a replaceable step for catalytic studies and artificial enzyme mimics provide an alternative way for catalysis and a better understanding of catalytic pathways at the same time. Here we designed an artificial catalyst model by decorating peptide nanofibers with a covalently conjugated catalytic triad sequence. Owing to the self-assembling nature of the peptide amphiphiles, multiple action units can be presented on the surface for enhanced catalytic performance. The designed catalyst has shown an enzyme-like kinetics profile with a significant substrate affinity. The cooperative action in between catalytic triad amino acids has shown improved catalytic activity in comparison to only the histidine-containing control group. Histidine is an irreplaceable contributor to catalytic action and this is an additional reason for control group selection. This new method based on the self-assembly of covalently conjugated action units offers a new platform for enzyme investigations and their further applications. -

Peptide Chemistry up to Its Present State
Appendix In this Appendix biographical sketches are compiled of many scientists who have made notable contributions to the development of peptide chemistry up to its present state. We have tried to consider names mainly connected with important events during the earlier periods of peptide history, but could not include all authors mentioned in the text of this book. This is particularly true for the more recent decades when the number of peptide chemists and biologists increased to such an extent that their enumeration would have gone beyond the scope of this Appendix. 250 Appendix Plate 8. Emil Abderhalden (1877-1950), Photo Plate 9. S. Akabori Leopoldina, Halle J Plate 10. Ernst Bayer Plate 11. Karel Blaha (1926-1988) Appendix 251 Plate 12. Max Brenner Plate 13. Hans Brockmann (1903-1988) Plate 14. Victor Bruckner (1900- 1980) Plate 15. Pehr V. Edman (1916- 1977) 252 Appendix Plate 16. Lyman C. Craig (1906-1974) Plate 17. Vittorio Erspamer Plate 18. Joseph S. Fruton, Biochemist and Historian Appendix 253 Plate 19. Rolf Geiger (1923-1988) Plate 20. Wolfgang Konig Plate 21. Dorothy Hodgkins Plate. 22. Franz Hofmeister (1850-1922), (Fischer, biograph. Lexikon) 254 Appendix Plate 23. The picture shows the late Professor 1.E. Jorpes (r.j and Professor V. Mutt during their favorite pastime in the archipelago on the Baltic near Stockholm Plate 24. Ephraim Katchalski (Katzir) Plate 25. Abraham Patchornik Appendix 255 Plate 26. P.G. Katsoyannis Plate 27. George W. Kenner (1922-1978) Plate 28. Edger Lederer (1908- 1988) Plate 29. Hennann Leuchs (1879-1945) 256 Appendix Plate 30. Choh Hao Li (1913-1987) Plate 31. -

Artificial Heme Enzymes for the Construction of Gold-Based
International Journal of Molecular Sciences Article Artificial Heme Enzymes for the Construction of Gold-Based Biomaterials Gerardo Zambrano 1 , Emmanuel Ruggiero 1,†, Anna Malafronte 1, Marco Chino 1 , Ornella Maglio 1,2 , Vincenzo Pavone 1, Flavia Nastri 1,* and Angela Lombardi 1,* 1 Department of Chemical Sciences, University of Napoli “Federico II” Via Cintia, 80126 Napoli, Italy; [email protected] (G.Z.); [email protected] (E.R.); [email protected] (A.M.); [email protected] (M.C.); [email protected] (O.M.); [email protected] (V.P.) 2 Istituto di Biostrutture e Bioimmagini, CNR, Via Mezzocannone 16, 80134 Napoli, Italy * Correspondence: fl[email protected] (F.N.); [email protected] (A.L.); Tel.: +39-081-674419 (F.N.); +39-081-674418 (A.L.) † Current address: BASF SE, Dept. Material Physics, Carl-Bosch-Strasse 38, 67056 Ludwigshafen, Germany. Received: 30 July 2018; Accepted: 19 September 2018; Published: 24 September 2018 Abstract: Many efforts are continuously devoted to the construction of hybrid biomaterials for specific applications, by immobilizing enzymes on different types of surfaces and/or nanomaterials. In addition, advances in computational, molecular and structural biology have led to a variety of strategies for designing and engineering artificial enzymes with defined catalytic properties. Here, we report the conjugation of an artificial heme enzyme (MIMO) with lipoic acid (LA) as a building block for the development of gold-based biomaterials. We show that the artificial MIMO@LA can be successfully conjugated to gold nanoparticles or immobilized onto gold electrode surfaces, displaying quasi-reversible redox properties and peroxidase activity. -

Abiological Catalysis by Artificial Haem Proteins Containing Noble Metals in Place of Iron Hanna M
LETTER doi:10.1038/nature17968 Abiological catalysis by artificial haem proteins containing noble metals in place of iron Hanna M. Key1,2*, Paweł Dydio1,2*, Douglas S. Clark3,4 & John F. Hartwig1,2 Enzymes that contain metal ions—that is, metalloenzymes— N–H and S–H bonds, but they do not catalyse the insertion into less possess the reactivity of a transition metal centre and the potential reactive C–H bonds3,14. of molecular evolution to modulate the reactivity and substrate- Because the repertoire of reactions catalysed by free metal-porphyrin selectivity of the system1. By exploiting substrate promiscuity complexes of Ru (ref. 15), Rh (ref. 16) and Ir (ref. 17) is much greater and protein engineering, the scope of reactions catalysed by than that of the free Fe-analogues, we hypothesized that their incorpora- native metalloenzymes has been expanded recently to include tion into PIX proteins could create new enzymes for abiological catalysis abiological transformations2,3. However, this strategy is limited by that is not possible with Fe-PIX enzymes. Artificial PIX proteins contain- the inherent reactivity of metal centres in native metalloenzymes. ing Mn, Cr, and Co cofactors have been prepared to mimic the intrin- To overcome this limitation, artificial metalloproteins have sic chemistry of the native haem proteins18–21, but the reactivities and been created by incorporating complete, noble-metal complexes selectivities of these processes are lower than those achieved in the same within proteins lacking native metal sites1,4,5. The interactions reactions catalysed by native Fe-PIX enzymes. Thus, artificially metal- of the substrate with the protein in these systems are, however, lated PIX proteins that catalyse reactions that are not catalysed by native distinct from those with the native protein because the metal Fe-PIX proteins are unknown, and the current, inefficient methods complex occupies the substrate binding site. -

Lecture 1: Strategies and Tactics in Organic Synthesis
Massachusetts Institute of Technology Organic Chemistry 5.511 October 3, 2007 Prof. Rick L. Danheiser Lecture 1: Strategies and Tactics in Organic Synthesis Retrosynthetic Analysis The key to the design of efficient syntheses ". the grand thing is to be able to reason backwards. That is a very useful accomplishment, and a very easy "The end is where we start from...." one, but people do not practice it much." T. S. Eliot, in "The Four Quartets" Sherlock Holmes, in "A Study in Scarlet" Strategy Tactics overall plan to achieve the means by which plan ultimate synthetic target is implemented intellectual experimental retrosynthetic planning synthetic execution TRANSFORMS REACTIONS Target Precursor Precursor Target Definitions Retron Structural unit that signals the application of a particular strategy algorithm during retrosynthetic analysis. Transform Imaginary retrosynthetic operation transforming a target molecule into a precursor molecule in a manner such that bond(s) can be reformed (or cleaved) by known or reasonable synthetic reactions. Strategy Algorithm Step-by-step instructions for performing a retrosynthetic operation. "...even in the earliest stages of the process of simplification of a synthetic problem, the chemist must make use of a particular form of analysis which depends on the interplay between structural features that exist in the target molecule and the types of reactions or synthetic operations available from organic chemistry for the modification or assemblage of structural units. The synthetic chemist has learned by experience to recognize within a target molecule certain units which can be synthesized, modified, or joined by known or conceivable synthetic operations...it is convenient to have a term for such units; the term "synthon" is suggested. -

Catalytically Active Nanomaterials: Artificial Enzymes of Next Generation Sanjay Singh*
www.symbiosisonline.org Symbiosis www.symbiosisonlinepublishing.com Research article Nanoscience & Technology: Open Access Open Access Catalytically Active Nanomaterials: Artificial Enzymes of Next Generation Sanjay Singh* Division of Biological and Life Sciences, School of Arts and Sciences, Ahmedabad University, Central Campus, Navrangpura, Ahmedabad 380009, Gujarat, India Received: November 17, 2017; Accepted: December 5, 2017; Published: December 10, 2017 *Corresponding author: Sanjay Singh, Division of Biological and Life Sciences, School of Arts and Sciences, Ahmedabad University, Central Campus, Navrangpura, Ahmedabad - 380009, Gujarat, India. Tel: +91-79-61911270. E-mail: [email protected] Abstract as they offer low cost of production, size, and shape mediated control over catalytic activity and high stability in extreme Due to the inherent limitations associated with natural enzymes, physiological conditions. Utilizing the enzymatic properties discovery and development of nanomaterials-based artificial enzymes of these nanozymes, several detection methods of analytes in (nanozymes) are highly desired. These nanozymes may address extremely low concentration has also been devised [7, 11-14]. the issues with practical applications of natural enzymes such as high cost of synthesis, purification, storage and limited spectrum Considering the above-mentioned advantages of nanozymes, of catalytic activity. In this review article, a crisp description of the enzymes.in this review article, we comprehensively discuss the recent recent progress in exploring, bio catalytic activity and constructing developments and future direction of nanomaterials as artificial nanozymes, including AuNPs, carbon-based nanomaterials, and CeO2 NPs are discussed. A brief description of new or enhanced applications of these nanozymes in bio diagnosis and therapeutics has also been Recent utilization of enzymes in the construction of provided. -
![[2.2]Paracyclophanes- Structure and Reactivity Studies Von Der](https://docslib.b-cdn.net/cover/2287/2-2-paracyclophanes-structure-and-reactivity-studies-von-der-1602287.webp)
[2.2]Paracyclophanes- Structure and Reactivity Studies Von Der
Syntheses of Functionalised [2.2]Paracyclophanes- Structure and Reactivity Studies Von der Gemeinsamen Naturwissenschaftlichen Fakultät der Technischen Universität Carolo Wilhelmina zu Braunschweig zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr.rer.nat.) genehmigte D i s s e r t a t i o n von Swaminathan Vijay Narayanan aus Chennai (Madras) / India 1. Referent: Prof. Dr. Dr. h. c. Henning Hopf 2. Referentin: Prof. Dr. Monika Mazik eingereicht am: 20. Jan 2005 mündliche Prüfung (Disputation) am: 29. März 2005 Druckjahr 2005 Vorveröffentlichungen der Dissertation Teilergebnisse aus dieser Arbeit wurden mit Genehmigung der Gemeinsamen Naturwissenschaft-lichen Fakultät, vertreten durch die Mentorin oder den Mentor/die Betreuerin oder den Betreuer der Arbeit, in folgenden Beiträgen vorab veröffentlicht: Publikationen K. El Shaieb, V. Narayanan, H. Hopf, I. Dix, A. Fischer, P. G. Jones, L. Ernst & K. Ibrom.: 4,15-Diamino[2.2]paracyclophane as a starting material for pseudo-geminally substituted [2.2]paracyclophanes. Eur. J. Org. Chem.: 567-577 (2003). Die vorliegende Arbeit wurde in der Zeit von Oktober 2001 bis Januar 2004 am Institut für Organische Chemie der Technischen Universität Braunschweig unter der Leitung von Prof. Dr. Dr. h.c. Henning Hopf angefertigt. It is my great pleasure to express my sincere gratitude to Prof. Dr. Henning Hopf for his support, encouragement and guidance throughout this research work. I thank him a lot for his invaluable ideas and remarks which made this study very interesting. I admire deep from my heart his energetic way of working and his brilliant ideas which made possible this research work to cover a vast area. -
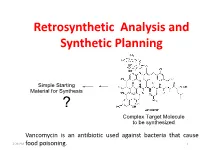
Retrosynthetic Analysis and Synthetic Planning
Retrosynthetic Analysis and Synthetic Planning Vancomycin is an antibiotic used against bacteria that cause 2:24 PM food poisoning. 1 Life’s Perspectives Planning a Journey to the Unknown 2:24 PM 2 Retrosynthetic Analysis Definition Retrosynthetic analysis (retrosynthesis) is a technique for planning a synthesis, especially of complex organic molecules, whereby the complex target molecule (TM) is reduced into a sequence of progressively simpler structures along a pathway which ultimately leads to the identification of a simple or commercially available starting material (SM) from which a chemical synthesis can then be developed. Retrosynthetic analysis is based on known reactions (e.g the Wittig reaction, oxidation, reduction etc). The synthetic plan generated from the retrosynthetic analysis will be the roadmap to guide the synthesis of the target molecule. 2:24 PM 3 Synthetic Planning Definition Synthesis is a construction process that involves converting simple or commercially available molecules into complex molecules using specific reagents associated with known reactions in the retrosynthetic scheme. Syntheses can be grouped into two broad categories: (i) Linear syntheses 2:24 PM (ii)Convergent syntheses 4 Linear Synthesis Definition In linear synthesis, the target molecule is synthesized through a series of linear transformations. Since the overall yield of the synthesis is based on the single longest route to the target molecule, by being long, a linear synthesis suffers a lower overall yield. The linear synthesis is fraught with failure for its lack of flexibility leading to potential large losses in the material already invested in the synthesis at the time of failure. 2:24 PM 5 Convergent Synthesis Definition In convergent synthesis, key fragments of the target molecule are synthesized separately or independently and then brought together at a later stage in the synthesis to make the target molecule.