HOXD8/DIAPH2-AS1 Epigenetically Regulates PAX3 and Impairs HTR-8/Svneo Cell Function Under Hypoxia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 7: Monogenic Forms of Diabetes
CHAPTER 7 MONOGENIC FORMS OF DIABETES Mark A. Sperling, MD, and Abhimanyu Garg, MD Dr. Mark A. Sperling is Emeritus Professor and Chair, University of Pittsburgh, Department of Pediatrics, Children’s Hospital of Pittsburgh of UPMC, Pittsburgh, PA. Dr. Abhimanyu Garg is Professor of Internal Medicine and Chief of the Division of Nutrition and Metabolic Diseases at University of Texas Southwestern Medical Center, Dallas, TX. SUMMARY Types 1 and 2 diabetes have multiple and complex genetic influences that interact with environmental triggers, such as viral infections or nutritional excesses, to result in their respective phenotypes: young, lean, and insulin-dependence for type 1 diabetes patients or older, overweight, and often manageable by lifestyle interventions and oral medications for type 2 diabetes patients. A small subset of patients, comprising ~2%–3% of all those diagnosed with diabetes, may have characteristics of either type 1 or type 2 diabetes but have single gene defects that interfere with insulin production, secretion, or action, resulting in clinical diabetes. These types of diabetes are known as MODY, originally defined as maturity-onset diabetes of youth, and severe early-onset forms, such as neonatal diabetes mellitus (NDM). Defects in genes involved in adipocyte development, differentiation, and death pathways cause lipodystrophy syndromes, which are also associated with insulin resistance and diabetes. Although these syndromes are considered rare, more awareness of these disorders and increased availability of genetic testing in clinical and research laboratories, as well as growing use of next generation, whole genome, or exome sequencing for clinically challenging phenotypes, are resulting in increased recognition. A correct diagnosis of MODY, NDM, or lipodystrophy syndromes has profound implications for treatment, genetic counseling, and prognosis. -

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -

The Title of the Dissertation
UNIVERSITY OF CALIFORNIA SAN DIEGO Novel network-based integrated analyses of multi-omics data reveal new insights into CD8+ T cell differentiation and mouse embryogenesis A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics and Systems Biology by Kai Zhang Committee in charge: Professor Wei Wang, Chair Professor Pavel Arkadjevich Pevzner, Co-Chair Professor Vineet Bafna Professor Cornelis Murre Professor Bing Ren 2018 Copyright Kai Zhang, 2018 All rights reserved. The dissertation of Kai Zhang is approved, and it is accept- able in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California San Diego 2018 iii EPIGRAPH The only true wisdom is in knowing you know nothing. —Socrates iv TABLE OF CONTENTS Signature Page ....................................... iii Epigraph ........................................... iv Table of Contents ...................................... v List of Figures ........................................ viii List of Tables ........................................ ix Acknowledgements ..................................... x Vita ............................................. xi Abstract of the Dissertation ................................. xii Chapter 1 General introduction ............................ 1 1.1 The applications of graph theory in bioinformatics ......... 1 1.2 Leveraging graphs to conduct integrated analyses .......... 4 1.3 References .............................. 6 Chapter 2 Systematic -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Evidence Revealing Deregulation of the KLF11-Mao a Pathway in Association with Chronic Stress and Depressive Disorders
Neuropsychopharmacology (2015) 40, 1373–1382 & 2015 American College of Neuropsychopharmacology. All rights reserved 0893-133X/15 www.neuropsychopharmacology.org Evidence Revealing Deregulation of The KLF11-Mao A Pathway in Association with Chronic Stress and Depressive Disorders 1 1 1,2 1 3 Sharonda Harris , Shakevia Johnson , Jeremy W Duncan , Chinelo Udemgba , Jeffrey H Meyer , Paul R Albert4, Gwen Lomberk5, Raul Urrutia5, Xiao-Ming Ou1, Craig A Stockmeier1,6 and Jun Ming Wang*,1,2,7 1 2 3 Department of Psychiatry and Human Behavior, Jackson, MS, USA; Program in Neuroscience, Jackson, MS, USA; Centre for Addiction and Mental Health and Department of Psychiatry, University of Toronto, Toronto, Ontario, Canada; 4Ottawa Hospital Research Institute (Neuroscience), Ottawa, Ontario, Canada; 5Epigenetics and Chromatin Dynamics Laboratory, GI Research Unit, Mayo Clinic, Rochester, MN, 6 7 USA; Department of Psychiatry, Case Western Reserve University, Cleveland, OH, USA; Department of Pathology, University of Mississippi Medical Center, Jackson, MS, USA The biochemical pathways underlying major depressive disorder (MDD) and chronic stress are not well understood. However, it has been reported that monoamine oxidase A (MAO A, a major neurotransmitter-degrading enzyme) is significantly increased in the brains of human subjects affected with MDD and rats exposed to chronic social defeat (CSD) stress, which is used to model depression. In the current study, we compared the protein levels of a MAO A-transcriptional activator, Kruppel-like factor 11 (KLF11 , also recognized as transforming growth factor-beta-inducible early gene 2) between the brains of 18 human subjects with MDD and 18 control subjects. We found that, indeed, the expression of KLF11 is increased by 36% (po0.02) in the postmortem prefrontal cortex of human subjects with MDD compared with controls. -

TWIST1 and TWIST2 Regulate Glycogen Storage and Inflammatory
J M MUDRY and others TWIST regulation of metabolism 224:3 303–313 Research in muscle TWIST1 and TWIST2 regulate glycogen storage and inflammatory genes in skeletal muscle Correspondence 1 1 2 1,2 Jonathan M Mudry , Julie Massart , Ferenc L M Szekeres and Anna Krook should be addressed to A Krook 1Section for Integrative Physiology, Department of Molecular Medicine and Surgery, and 2Section for Integrative Email Physiology, Department of Physiology and Pharmacology, Karolinska Institutet, SE-171 77 Stockholm, Sweden [email protected] Abstract TWIST proteins are important for development of embryonic skeletal muscle and play a role Key Words in the metabolism of tumor and white adipose tissue. The impact of TWIST on metabolism " TWIST in skeletal muscle is incompletely studied. Our aim was to assess the impact of TWIST1 " metabolism and TWIST2 overexpression on glucose and lipid metabolism. In intact mouse muscle, " glycogen overexpression of Twist reduced total glycogen content without altering glucose uptake. " skeletal muscle Expression of TWIST1 or TWIST2 reduced Pdk4 mRNA, while increasing mRNA levels of Il6, Tnfa, and Il1b. Phosphorylation of AKT was increased and protein abundance of acetyl CoA carboxylase (ACC) was decreased in skeletal muscle overexpressing TWIST1 or TWIST2. Glycogen synthesis and fatty acid oxidation remained stable in C2C12 cells overexpressing TWIST1 or TWIST2. Finally, skeletal muscle mRNA levels remain unaltered in ob/ob mice, type 2 diabetic patients, or in healthy subjects before and after 3 months of exercise training. Journal of Endocrinology Collectively, our results indicate that TWIST1 and TWIST2 are expressed in skeletal muscle. Overexpression of these proteins impacts proteins in metabolic pathways and mRNA level of cytokines. -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals
animals Review Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals Mohammadreza Mohammadabadi 1 , Farhad Bordbar 1,* , Just Jensen 2 , Min Du 3 and Wei Guo 4 1 Department of Animal Science, Faculty of Agriculture, Shahid Bahonar University of Kerman, Kerman 77951, Iran; [email protected] 2 Center for Quantitative Genetics and Genomics, Aarhus University, 8210 Aarhus, Denmark; [email protected] 3 Washington Center for Muscle Biology, Department of Animal Sciences, Washington State University, Pullman, WA 99163, USA; [email protected] 4 Muscle Biology and Animal Biologics, Animal and Dairy Science, University of Wisconsin-Madison, Madison, WI 53558, USA; [email protected] * Correspondence: [email protected] Simple Summary: Skeletal muscle mass is an important economic trait, and muscle development and growth is a crucial factor to supply enough meat for human consumption. Thus, understanding (candidate) genes regulating skeletal muscle development is crucial for understanding molecular genetic regulation of muscle growth and can be benefit the meat industry toward the goal of in- creasing meat yields. During the past years, significant progress has been made for understanding these mechanisms, and thus, we decided to write a comprehensive review covering regulators and (candidate) genes crucial for muscle development and growth in farm animals. Detection of these genes and factors increases our understanding of muscle growth and development and is a great help for breeders to satisfy demands for meat production on a global scale. Citation: Mohammadabadi, M.; Abstract: Farm-animal species play crucial roles in satisfying demands for meat on a global scale, Bordbar, F.; Jensen, J.; Du, M.; Guo, W. -
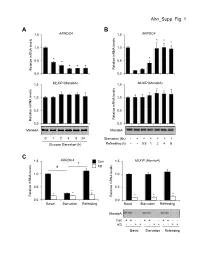
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

High ELF4 Expression in Human Cancers Is Associated with Worse Disease Outcomes and Increased Resistance to Anticancer Agents
medRxiv preprint doi: https://doi.org/10.1101/2020.05.16.20104380; this version posted May 20, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . High ELF4 Expression in Human Cancers is Associated with Worse Disease Outcomes and Increased Resistance to Anticancer Agents Doris Kafita1φ, Victor Daka4φ, Panji Nkhoma1φ, Mildred Zulu3, Ephraim Zulu1, Rabecca Tembo3, Zifa Ngwira3, Florence Mwaba3, Musalula Sinkala1, Sody Munsaka1 1, University of Zambia, School of Health Sciences, Department of Biomedical Sciences, P.O. Box 50110, Nationalist Road, Lusaka, Zambia 2, University of Zambia, School of Medicine, Department of Pathology and Microbiology, P.O. Box 50110, Nationalist Road, Lusaka, Zambia 3, Copperbelt University, School of Medicine, Ndola, Zambia φ, Contributed equally Correspondence [email protected] [email protected] NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.05.16.20104380; this version posted May 20, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . Abstract The malignant phenotype of tumour cells is fuelled by changes in the expression of various transcription factors, which include some of the well-studied proteins such as p53 and Myc. -

Genome-Wide Identification of the Early Flowering 4 (ELF4) Gene Family in Cotton and Silent Ghelf4-1 and Ghefl3-6 Decreased Cott
fgene-12-686852 July 13, 2021 Time: 16:58 # 1 ORIGINAL RESEARCH published: 06 July 2021 doi: 10.3389/fgene.2021.686852 Genome-Wide Identification of the Early Flowering 4 (ELF4) Gene Family in Cotton and Silent GhELF4-1 and GhEFL3-6 Decreased Cotton Stress Resistance Miaomiao Tian1,2, Aimin Wu2, Meng Zhang2, Jingjing Zhang2, Hengling Wei2, Xu Yang2, Liang Ma2, Jianhua Lu2, Xiaokang Fu2, Hantao Wang2* and Shuxun Yu1,2* 1 Engineering Research Centre of Cotton, Ministry of Education, College of Agriculture, Xinjiang Agricultural University, Edited by: Ürümqi, China, 2 State Key Laboratory of Cotton Biology, Institute of Cotton Research, Chinese Academy of Agricultural Zefeng Yang, Sciences, Anyang, China Yangzhou University, China Reviewed by: The early flowering 4 (ELF4) family members play multiple roles in the physiological Waqas Shafqat Chattha, University of Agriculture, Faisalabad, development of plants. ELF4s participated in the plant biological clock’s regulation Pakistan process, photoperiod, hypocotyl elongation, and flowering time. However, the function Ying Bao, in the ELF4s gene is barely known. In this study, 11, 12, 21, and 22 ELF4 genes Qufu Normal University, China Yijun Wang, were identified from the genomes of Gossypium arboreum, Gossypium raimondii, Yangzhou University, China Gossypium hirsutum, and Gossypium barbadense, respectively. There ELF4s genes *Correspondence: were classified into four subfamilies, and members from the same subfamily show Hantao Wang [email protected] relatively conservative gene structures. The results of gene chromosome location and Shuxun Yu gene duplication revealed that segmental duplication promotes gene expansion, and the [email protected] Ka/Ks indicated that the ELF4 gene family has undergone purification selection during Specialty section: long-term evolution.