Direct Analysis in Real Time-Mass Spectrometry for Rapid Quantification
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
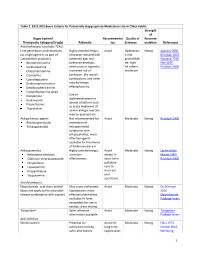
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Antiparasitic Properties of Cardiovascular Agents Against Human Intravascular Parasite Schistosoma Mansoni
pharmaceuticals Article Antiparasitic Properties of Cardiovascular Agents against Human Intravascular Parasite Schistosoma mansoni Raquel Porto 1, Ana C. Mengarda 1, Rayssa A. Cajas 1, Maria C. Salvadori 2 , Fernanda S. Teixeira 2 , Daniel D. R. Arcanjo 3 , Abolghasem Siyadatpanah 4, Maria de Lourdes Pereira 5 , Polrat Wilairatana 6,* and Josué de Moraes 1,* 1 Research Center for Neglected Diseases, Guarulhos University, Praça Tereza Cristina 229, São Paulo 07023-070, SP, Brazil; [email protected] (R.P.); [email protected] (A.C.M.); [email protected] (R.A.C.) 2 Institute of Physics, University of São Paulo, São Paulo 05508-060, SP, Brazil; [email protected] (M.C.S.); [email protected] (F.S.T.) 3 Department of Biophysics and Physiology, Federal University of Piaui, Teresina 64049-550, PI, Brazil; [email protected] 4 Ferdows School of Paramedical and Health, Birjand University of Medical Sciences, Birjand 9717853577, Iran; [email protected] 5 CICECO-Aveiro Institute of Materials & Department of Medical Sciences, University of Aveiro, 3810-193 Aveiro, Portugal; [email protected] 6 Department of Clinical Tropical Medicine, Faculty of Tropical Medicine, Mahidol University, Bangkok 10400, Thailand * Correspondence: [email protected] (P.W.); [email protected] (J.d.M.) Citation: Porto, R.; Mengarda, A.C.; Abstract: The intravascular parasitic worm Schistosoma mansoni is a causative agent of schistosomiasis, Cajas, R.A.; Salvadori, M.C.; Teixeira, a disease of great global public health significance. Praziquantel is the only drug available to F.S.; Arcanjo, D.D.R.; Siyadatpanah, treat schistosomiasis and there is an urgent demand for new anthelmintic agents. -

Multaq, INN-Dronedarone
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT MULTAQ 400 mg film-coated tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each tablet contains 400 mg of dronedarone (as hydrochloride). Excipient with known effect: Each tablet also contains 41.65 mg of lactose (as monohydrate). For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Film-coated tablet (tablet). White, oblong shaped tablets, engraved with a double wave marking on one side and “4142”code on the other side. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications MULTAQ is indicated for the maintenance of sinus rhythm after successful cardioversion in adult clinically stable patients with paroxysmal or persistent atrial fibrillation (AF). Due to its safety profile (see sections 4.3 and 4.4), MULTAQ should only be prescribed after alternative treatment options have been considered. MULTAQ must not be given to patients with left ventricular systolic dysfunction or to patients with current or previous episodes of heart failure. 4.2 Posology and method of administration Treatment should be initiated and monitored only under specialist supervision (see section 4.4). Treatment with dronedarone can be initiated in an outpatient setting. Treatment with Class I or III antiarrhythmics (such as flecainide, propafenone, quinidine, disopyramide, dofetilide, sotalol, amiodarone) must be stopped before starting dronedarone. There is limited information on the optimal timing to switch from amiodarone to dronedarone. It should be considered that amiodarone may have a long duration of action after discontinuation due to its long half-life. If a switch is envisaged, this should be done under the supervision of a specialist (see sections 4.3 and 5.1). -

EGSC Drug and Alcohol Policy
EGSC Student Handbook, “Institutional Policy” section Revisions adopted by the President’s Cabinet 9/22/15 Approved by President’s Cabinet 9-23-14 Drugs and Alcohol President Bush’s National Drug Control Strategy issued in September of 1989 proposed that the Congress pass legislation to require schools, colleges, and universities to implement and enforce firm drug prevention programs and policies. On December 12, 1989, the President signed the Drug-Free Schools and Communities Act Amendments (Public Law 101-226). This law establishes a process to accomplish the President’s goal. Public Law 101-226 requires that schools maintain standards of conduct that clearly prohibit, at a minimum, the unlawful possession, use, or distribution of drugs and alcohol on school property or as any part of school activities. In support of Public Law 101-226, East Georgia State College recognizes and supports local, state, and federal laws with respect to the sale, use, distribution, and possession of alcoholic beverages and illegal drugs. To this end, the possession, consumption, distribution, manufacture, or sale (without valid medical or dental prescription) of alcoholic beverages, illegal or dangerous drugs on East Georgia State College property or at institutionally-approved events off campus is prohibited. East Georgia State College also adheres to the following: Drug Free School Zone – it is unlawful for persons to manufacture, distribute, dispense, or possess with intent to distribute illegal drugs within 1,000 feet of any elementary or secondary school property. A first conviction is punishable by imprisonment for not more than 20 years and/or a fine of not more than $20,000, and a second or subsequent conviction is punishable by imprisonment for at least five years but not more than 40 years and/or a fine of not more than $40,000. -

MULTAQ (Dronedarone) Tablets Suspension Or Discontinuation of MULTAQ (5.1) Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION • Pregnancy (4, 8.1) These highlights do not include all the information needed to use • Nursing mothers (4, 8.3) MULTAQ safely and effectively. See full prescribing information for MULTAQ. -----------------------WARNINGS AND PRECAUTIONS------------------------ • Heart failure: If heart failure develops or worsens, consider the MULTAQ (dronedarone) Tablets suspension or discontinuation of MULTAQ (5.1) Initial U.S. Approval: 2009 • Hypokalemia and hypomagnesemia: Maintain potassium and magnesium levels within the normal range (5.2) WARNING: HEART FAILURE • QT prolongation: Stop MULTAQ if QTc Bazett ≥500ms (5.3) MULTAQ is contraindicated in patients with NYHA Class IV heart • Increase in creatinine: Within a week, MULTAQ causes a small increase failure or NYHA Class II - III heart failure with a recent in serum creatinine that does not reflect a change in underlying renal decompensation requiring hospitalization or referral to a specialized function (5.4) heart failure clinic (4). • Teratogen: Women of childbearing potential should use effective contraception while using MULTAQ (5.5) In a placebo-controlled study in patients with severe heart failure requiring recent hospitalization or referral to a specialized heart failure ------------------------------ADVERSE REACTIONS------------------------------- clinic for worsening symptoms (the ANDROMEDA Study), patients Most common adverse reactions (≥2%) are diarrhea, nausea, abdominal pain, given dronedarone had a greater than two-fold increase in mortality. -

Oral Antiarrhythmic Drugs in Converting Recent Onset Atrial Fibrillation
Review article Oral antiarrhythmic drugs in converting recent onset atrial fibrillation • Vera H.M. Deneer, Marieke B.I. Borgh, J. Herre Kingma, Loraine Lie-A-Huen and Jacobus R.B.J. Brouwers Introduction Pharm World Sci 2004; 26: 66–78. © 2004 Kluwer Academic Publishers. Printed in the Netherlands. Atrial fibrillation is the most common arrhythmia. The incidence of atrial fibrillation depends on the age of V.H.M. Deneer (correspondence, e-mail: the study population. The incidence varies between 2 [email protected]), M.B.I. Borgh, L. Lie-A-Huen: Department of Clinical Pharmacy or 3 new cases per 1,000 population per year between J.H. Kingma: Department of Cardiology, St Antonius Hospital, the ages of 55 and 64 years to 35 new cases per 1,000 Koekoekslaan 1, 3435 CM Nieuwegein, The Netherlands population per year between the ages of 85 and 94 J.R.B.J. Brouwers: Groningen University Institute for Drug 1 Exploration (GUIDE), Section of Pharmacotherapy, University years . Treatment of an episode of paroxysmal atrial fi- of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, brillation consists of restoring sinus rhythm by DC- The Netherlands electrical cardioversion or by the intravenous adminis- Key words tration of an antiarrhythmic drug, but frequently the Atrial fibrillation arrhythmia spontaneously terminates1–3. After one or Amiodarone more episodes of atrial fibrillation chronic prophylac- Antiarrhythmic drugs Digoxin tic treatment with an antiarrhythmic drug is often Episodic treatment started for maintenance of sinus rhythm4–8. Another Flecainide treatment strategy consists of allowing the arrhythmia Propafenone Quinidine to exist in combination with pharmacological ven- Sotalol tricular rate control. -

Severe Cardiac Toxicity Following Alcohol Intake in a Patient Using Therapeutic Dose of Propafenone
752 Turk Kardiyol Dern Ars 2017;45(8):752-754 doi: 10.5543/tkda.2017.84553 CASE REPORT Severe cardiac toxicity following alcohol intake in a patient using therapeutic dose of propafenone Terapötik dozda propafenon kullanmakta olan bir hastada alkol kullanımı sonrası gelişen ciddi kardiyak toksisite Zübeyde Bayram, M.D., Ahmet Güner, M.D., Cem Doğan, M.D., Fatih Yılmaz, M.D., Nihal Özdemir, M.D. Department of Cardiology, Kartal Koşuyolu Yüksek İhtisas Training and Research Hospital, İstanbul, Turkey Summary– An unconscious, 25-year-old, male patient Özet– Yirmi beş yaşında erkek hasta acil servise bilincini was brought to the emergency department. The patient’s yitirmiş olarak getirildi. Hastanın elektrokardiyografisinde electrocardiography demonstrated a wide QRS interval QRS genişlemesi ve birinci derece atriyoventriküler blok and first-degree atrioventricular block. He was being treat- görüldü. Hasta nöbetli atriyum fibrilasyonu nedeniyle gün- ed with propafenone twice daily (450 mg) for paroxysmal de iki kez propafenon kullanıyormuş (450 mg). Hastanın bir atrial fibrillation. The patient had consumed a substantial gün önce alkol kullanımı öyküsü de mevcuttu. Hasta sod- amount of alcohol the day before. He recovered after sup- yum bikarbonat ve inotrop destekleyici tedavi sonrasında portive management with sodium bicarbonate and inotropic iyileşti. Sunulan bu olguda, uygulanan tedavi sonucunda therapy. In the presently described case, treatment resulted QRS genişlemesinin ve hemodiamik durumun düzelmesini in quick normalization of QRS interval and stabilization of sağlandı. hemodynamic status. ropafenone is a Vaughan Williams class I anti- raphy revealed an ejec- Abbreviations: Parrhythmic agent. It is used in the treatment of tion fraction of 55%. CPR Cardiopulmonary resuscitation supraventricular tachyarrhythmias. -

Cardioversion of AF the Pill-In-The-Pocket
Cardioversion of AF the pill-in-the-pocket J Mansourati Brest - France NO CONFLICT OF INTEREST TO DECLARE 532 Bunch and Gersh: Rhythm Control Strategies and the Role of Antiarrhythmic Drugs JGIM Figure 1. Broad consequences of atrial fibrillation. that patient characteristics such as age and concomitant more difficult it becomes to restore normal sinus rhythm, likely conditions such as CHF influence clinical outcomes. For because the electrical, contractile, and structural changes that example, subset analysis of AFFIRM demonstrated that create an optimal environment for AF propagation.23 These younger patients (≤65 years of age) and patients without a structural observations with durable AF imply a question: does history of CHF benefited more (had better survival) from rate aggressive upfront restoration of sinus rhythm with either versus rhythm control therapy.17 These trials highlight the nonpharmacologic or pharmacologic interventions improve relative risks of AF itself when compared to antiarrhythmic outcomes?23,24 therapy. In this commentary, we address the pathologic The potential consequences of AF and AF-associated remo- mechanisms of AF, current trials that investigate rate and deling are significant (Fig. 1). The hemodynamic consequences rhythm strategies, and future therapies that may change of atrial fibrillation may give rise to symptoms (e.g., discomfort, treatment approaches based on preliminary evidence suggest- palpitations, breathlessness, syncope, dizziness, reduced ex- ing a more favorable safety profile. ercise tolerance, -

Bicarbonate Therapy for Unstable Propafenone-Induced Wide Complex Tachycardia
PHARMACOTHERAPY • PHARMACOTHÉRAPIE Bicarbonate therapy for unstable propafenone-induced wide complex tachycardia Jeff Brubacher, MD ABSTRACT A previously healthy 73-year-old woman presented to hospital with acute atrial fibrillation. After intravenous procainamide failed to restore sinus rhythm, she was treated with 300 mg of oral propafenone and discharged with a prescription for propafenone and propranolol. Six hours later she took 150 mg of propafenone as prescribed. Within 1 hour she became dyspneic and collapsed. On arrival in hospital she was unconscious, with a wide complex tachycardia and no obtainable blood pressure. After defibrillation and lidocaine, she converted to a wide complex sinus rhythm, but remained profoundly hypotensive despite intravenous epinephrine and dopamine. Hypertonic sodium bicarbonate (HCO3) was administered and, shortly thereafter, her blood pressure in- creased, her QRS duration normalized and her clinical status improved dramatically. In this case of severe refractory propafenone-related cardiac toxicity, intravenous HCO3 led to a profound clinical improvement. Emergency physicians should be familiar with the syndrome of sodium-channel blocker poisoning and recognize the potentially important role of bicarbonate in its treatment. Key words: propafenone; sodium channel blockers; anti-arrhythmia agents; adverse effects; atrial fibrillation RÉSUMÉ Une femme âgée de 73 ans auparavant en bonne santé s’est rendue à l’hôpital pour une fibrilla- tion auriculaire aiguë. La procaïnamide intraveineuse n’ayant pas réussi à rétablir le rythme si- nusal, la patiente reçut 300 mg de propafénone per os et fut renvoyée chez elle avec une ordon- nance pour de la propafénone et du propranolol. Six heures plus tard, elle prit 150 mg de propafénone tel que prescrit. -

Data Sheet Rytmonorm
NEW ZEALAND DATA SHEET RYTMONORM® 1. Product Name Rytmonorm, 150 mg, film coated tablets. 2. Qualitative and Quantitative Composition Each Rytmonorm film coated tablet contains 150 mg of propafenone (as propafenone hydrochloride). For the full list of excipients, see section 6.1. 3. Pharmaceutical Form Rytmonorm 150 mg - white to off-white, round, biconvex film coated tablets embossed on one face with “150”. The other face is unmarked. 4. Clinical Particulars 4.1 Therapeutic indications For the treatment of supraventricular extrasystoles and supraventricular tachycardias, and in Wolff- Parkinson-White (WPW) syndrome. Also for the treatment of life-threatening documented ventricular tachyarrhythmia. The use of Rytmonorm is not recommended in patients with less severe ventricular arrhythmias, even if the patients are symptomatic. Because of the proarrhythmic effects of Rytmonorm, its use should be reserved for patients in whom, in the opinion of the physician, the potential benefits of treatment outweigh the risks. 4.2 Dose and method of administration Dose The use of Rytmonorm should be initiated by a specialist physician. The individual maintenance dose should be determined under cardiological surveillance including repeated ECG monitoring and repeated blood pressure control (titration phase). If the QRS interval is widened by more than 20%, the corrected QT interval is lengthened or second or third degree AV block occurs, the dose should be reduced or discontinued until the ECG returns to normal. The dosage is to be adjusted to the individual patient’s requirements. It is essential that each patient given Rytmonorm be evaluated electrocardiographically and clinically prior to and during therapy to determine whether the response to Rytmonorm supports continued treatment. -

Living with Atrial Fibrillation (Afib): Introduction
LIVING WITH ATRIAL FIBRILLATION Patient Guide University of North Carolina Department of Cardiology and Electrophysiology 1 Living with Atrial Fibrillation TABLE OF CONTENTS Living with Atrial Fibrillation (Afib): Introduction ......................................................... 3 Overview of Atrial Fibrillation .......................................................................................... 5 Who gets Afib? ...........................................................................................................................7 Types of Afib- What Type Do You Have? ................................................................................7 What Are Symptoms of Afib? ...................................................................................................8 What are the Dangers of Afib? ..................................................................................................8 Diagnosis of Afib ........................................................................................................................9 Afib Treatment .......................................................................................................................... 10 Important Points If You Are Taking Blood Thinners ............................................................ 11 Common Medications For Heart Rate Control in Afib ......................................................... 13 Getting The Heart back In Normal Rhythm: .......................................................................... 14 Antiarrhythmic -

Pharmacological Drugs Inducing Ototoxicity, Vestibular Symptoms and Tinnitus: a Reasoned and Updated Guide
European Review for Medical and Pharmacological Sciences 2011; 15: 601-636 Pharmacological drugs inducing ototoxicity, vestibular symptoms and tinnitus: a reasoned and updated guide G. CIANFRONE1, D. PENTANGELO1, F. CIANFRONE2, F. MAZZEI1, R. TURCHETTA1, M.P. ORLANDO1, G. ALTISSIMI1 1Department of Otolaryngology, Audiology and Phoniatrics, “Umberto I” University Hospital, Sapienza University, Rome (Italy); 2Institute of Otorhinolaryngology, School of Medicine, Catholic University of the Sacred Heart, Rome (Italy) Abstract. – The present work on drug-in- Introduction duced ototoxicity, tinnitus and vertigo repre- sents the update and revision of a previous The panorama of the pharmacological origin guide to adverse drug reactions for italian physi- iatrogenic noxae able to induce either harmful cians (2005). The panorama of drug-induced side effects causing ototoxicity or symptoms ototoxic effects or just a symptomatology like tin- such as tinnitus or dizziness and vertigo has en- nitus or balance disturbances, without any harm- larged in recent years, thanks to a better knowl- ful consequence, has widened in the last few edge and a more specific attention of pharma- years. The reason for this is the progress of scien- ceutical firms and drug-control institutions. In tific knowledge, the increased awareness of the daily clinical practice, there is a need for the pharmaceutical companies and of the institutions, family physician and the ENT specialist or audi- ologist (also in consideration of the possible which supervise pharmaceutical production. medico-legal implications) to focus the attention Only through continuous updating and experi- on the possible risk of otological side effects. ence sharing it’s possible to offer patients the This would allow a clinical risk-benefit evalua- certainty of receiving the treatment that is appro- tion, weighing the possible clinical advantage in priate, safe and effective and based upon the their field of competence against possible oto- most credited clinical studies.