Sarcomeric Deficits Underlie MYBPC1-Associated Myopathy with Myogenic Tremor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome Analysis and Knowledge
Dahary et al. BMC Medical Genomics (2019) 12:200 https://doi.org/10.1186/s12920-019-0647-8 SOFTWARE Open Access Genome analysis and knowledge-driven variant interpretation with TGex Dvir Dahary1*, Yaron Golan1, Yaron Mazor1, Ofer Zelig1, Ruth Barshir2, Michal Twik2, Tsippi Iny Stein2, Guy Rosner3,4, Revital Kariv3,4, Fei Chen5, Qiang Zhang5, Yiping Shen5,6,7, Marilyn Safran2, Doron Lancet2* and Simon Fishilevich2* Abstract Background: The clinical genetics revolution ushers in great opportunities, accompanied by significant challenges. The fundamental mission in clinical genetics is to analyze genomes, and to identify the most relevant genetic variations underlying a patient’s phenotypes and symptoms. The adoption of Whole Genome Sequencing requires novel capacities for interpretation of non-coding variants. Results: We present TGex, the Translational Genomics expert, a novel genome variation analysis and interpretation platform, with remarkable exome analysis capacities and a pioneering approach of non-coding variants interpretation. TGex’s main strength is combining state-of-the-art variant filtering with knowledge-driven analysis made possible by VarElect, our highly effective gene-phenotype interpretation tool. VarElect leverages the widely used GeneCards knowledgebase, which integrates information from > 150 automatically-mined data sources. Access to such a comprehensive data compendium also facilitates TGex’s broad variant annotation, supporting evidence exploration, and decision making. TGex has an interactive, user-friendly, and easy adaptive interface, ACMG compliance, and an automated reporting system. Beyond comprehensive whole exome sequence capabilities, TGex encompasses innovative non-coding variants interpretation, towards the goal of maximal exploitation of whole genome sequence analyses in the clinical genetics practice. This is enabled by GeneCards’ recently developed GeneHancer, a novel integrative and fully annotated database of human enhancers and promoters. -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways
biomolecules Article Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways Aayushi Srivastava 1,2,3,4 , Abhishek Kumar 1,5,6 , Sara Giangiobbe 1, Elena Bonora 7, Kari Hemminki 1, Asta Försti 1,2,3 and Obul Reddy Bandapalli 1,2,3,* 1 Division of Molecular Genetic Epidemiology, German Cancer Research Center (DKFZ), D-69120 Heidelberg, Germany; [email protected] (A.S.); [email protected] (A.K.); [email protected] (S.G.); [email protected] (K.H.); [email protected] (A.F.) 2 Hopp Children’s Cancer Center (KiTZ), D-69120 Heidelberg, Germany 3 Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ), German Cancer Consortium (DKTK), D-69120 Heidelberg, Germany 4 Medical Faculty, Heidelberg University, D-69120 Heidelberg, Germany 5 Institute of Bioinformatics, International Technology Park, Bangalore 560066, India 6 Manipal Academy of Higher Education (MAHE), Manipal, Karnataka 576104, India 7 S.Orsola-Malphigi Hospital, Unit of Medical Genetics, 40138 Bologna, Italy; [email protected] * Correspondence: [email protected]; Tel.: +49-6221-42-1709 Received: 29 August 2019; Accepted: 10 October 2019; Published: 13 October 2019 Abstract: Evidence of familial inheritance in non-medullary thyroid cancer (NMTC) has accumulated over the last few decades. However, known variants account for a very small percentage of the genetic burden. Here, we focused on the identification of common pathways and networks enriched in NMTC families to better understand its pathogenesis with the final aim of identifying one novel high/moderate-penetrance germline predisposition variant segregating with the disease in each studied family. -

Mouse Mybpc1 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Mybpc1 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Mybpc1 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Mybpc1 gene (NCBI Reference Sequence: NM_001252372 ; Ensembl: ENSMUSG00000020061 ) is located on Mouse chromosome 10. 29 exons are identified, with the ATG start codon in exon 1 and the TAG stop codon in exon 28 (Transcript: ENSMUST00000119185). Exon 3~4 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Mybpc1 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-408J20 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 3 starts from about 1.83% of the coding region. The knockout of Exon 3~4 will result in frameshift of the gene. The size of intron 2 for 5'-loxP site insertion: 12130 bp, and the size of intron 4 for 3'-loxP site insertion: 863 bp. The size of effective cKO region: ~2510 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 3 4 5 29 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Mybpc1 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
www.nature.com/scientificreports OPEN Assessment of a complete and classifed platelet proteome from genome‑wide transcripts of human platelets and megakaryocytes covering platelet functions Jingnan Huang1,2*, Frauke Swieringa1,2,9, Fiorella A. Solari2,9, Isabella Provenzale1, Luigi Grassi3, Ilaria De Simone1, Constance C. F. M. J. Baaten1,4, Rachel Cavill5, Albert Sickmann2,6,7,9, Mattia Frontini3,8,9 & Johan W. M. Heemskerk1,9* Novel platelet and megakaryocyte transcriptome analysis allows prediction of the full or theoretical proteome of a representative human platelet. Here, we integrated the established platelet proteomes from six cohorts of healthy subjects, encompassing 5.2 k proteins, with two novel genome‑wide transcriptomes (57.8 k mRNAs). For 14.8 k protein‑coding transcripts, we assigned the proteins to 21 UniProt‑based classes, based on their preferential intracellular localization and presumed function. This classifed transcriptome‑proteome profle of platelets revealed: (i) Absence of 37.2 k genome‑ wide transcripts. (ii) High quantitative similarity of platelet and megakaryocyte transcriptomes (R = 0.75) for 14.8 k protein‑coding genes, but not for 3.8 k RNA genes or 1.9 k pseudogenes (R = 0.43–0.54), suggesting redistribution of mRNAs upon platelet shedding from megakaryocytes. (iii) Copy numbers of 3.5 k proteins that were restricted in size by the corresponding transcript levels (iv) Near complete coverage of identifed proteins in the relevant transcriptome (log2fpkm > 0.20) except for plasma‑derived secretory proteins, pointing to adhesion and uptake of such proteins. (v) Underrepresentation in the identifed proteome of nuclear‑related, membrane and signaling proteins, as well proteins with low‑level transcripts. -
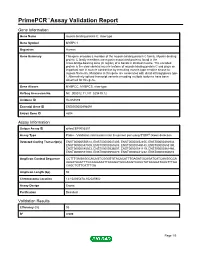
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name myosin binding protein C, slow type Gene Symbol MYBPC1 Organism Human Gene Summary This gene encodes a member of the myosin-binding protein C family. Myosin-binding protein C family members are myosin-associated proteins found in the cross-bridge-bearing zone (C region) of A bands in striated muscle. The encoded protein is the slow skeletal muscle isoform of myosin-binding protein C and plays an important role in muscle contraction by recruiting muscle-type creatine kinase to myosin filaments. Mutations in this gene are associated with distal arthrogryposis type I. Alternatively spliced transcript variants encoding multiple isoforms have been observed for this gene. Gene Aliases MYBPCC, MYBPCS, slow-type RefSeq Accession No. NC_000012.11, NT_029419.12 UniGene ID Hs.654589 Ensembl Gene ID ENSG00000196091 Entrez Gene ID 4604 Assay Information Unique Assay ID qHsaCEP0055301 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000550514, ENST00000547405, ENST00000452455, ENST00000392934, ENST00000547509, ENST00000361685, ENST00000549145, ENST00000553190, ENST00000545503, ENST00000536007, ENST00000541119, ENST00000361466, ENST00000551300, ENST00000550270, ENST00000441232, ENST00000360610 Amplicon Context Sequence CCTTTGAGAGGCACAGTCGGGTGTACACATTTGAGATGCAGATCATCAAGGCCA AAGATAACTTTGCAGGAAATTACAGATGCGAGGTCACCTATAAGGATAAGTTTGA CAGCTGTTCATTTGA Amplicon Length (bp) 94 Chromosome Location 12:102025478-102025902 Assay -

Comparative Gene Expression Profiling Between Human Cultured
Raymond et al. BMC Genomics 2010, 11:125 http://www.biomedcentral.com/1471-2164/11/125 RESEARCH ARTICLE Open Access Comparative gene expression profiling between human cultured myotubes and skeletal muscle tissue Frederic Raymond1, Sylviane Métairon1, Martin Kussmann1, Jaume Colomer2, Andres Nascimento2, Emma Mormeneo3, Cèlia García-Martínez3, Anna M Gómez-Foix3* Abstract Background: A high-sensitivity DNA microarray platform requiring nanograms of RNA input facilitates the application of transcriptome analysis to individual skeletal muscle (SM) tissue samples. Culturing myotubes from SM-biopsies enables investigating transcriptional defects and assaying therapeutic strategies. This study compares the transcriptome of aneurally cultured human SM cells versus that of tissue biopsies. Results: We used the Illumina expression BeadChips to determine the transcriptomic differences between tissue and cultured SM samples from five individuals. Changes in the expression of several genes were confirmed by QuantiGene Plex assay or reverse transcription real-time PCR. In cultured myotubes compared to the tissue, 1216 genes were regulated: 583 down and 633 up. Gene ontology analysis showed that downregulated genes were mainly associated with cytoplasm, particularly mitochondria, and involved in metabolism and the muscle-system/ contraction process. Upregulated genes were predominantly related to cytoplasm, endoplasmic reticulum, and extracellular matrix. The most significantly regulated pathway was mitochondrial dysfunction. Apoptosis genes were also modulated. Among the most downregulated genes detected in this study were genes encoding metabolic proteins AMPD1, PYGM, CPT1B and UCP3, muscle-system proteins TMOD4, MYBPC1, MYOZ1 and XIRP2, the proteolytic CAPN3 and the myogenic regulator MYF6. Coordinated reduced expression of five members of the GIMAP gene family, which form a cluster on chromosome 7, was shown, and the GIMAP4-reduction was validated. -

An En Masse Phenotype and Function Prediction System for Mus Musculus
Open Access Research2008TaetVolume al.s¸an 9, Suppl 1, Article S8 An en masse phenotype and function prediction system for Mus musculus Murat Taşan¤*, Weidong Tian¤*, David P Hill†, Francis D Gibbons‡, Judith A Blake† and Frederick P Roth* Addresses: *Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Longwood Avenue, Boston, Massachusetts 02115, USA. †Computational Biology and Bioinformatics, The Jackson Laboratory, Main Street, Bar Harbor, Maine 04609, USA. ‡Merrimack Pharmaceuticals, Kendall Square, Cambridge, Massachusetts 02139, USA. ¤ These authors contributed equally to this work. Correspondence: Frederick P Roth. Email: [email protected] Published: 27 June 2008 Genome Biology 2008, 9:S8 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2008/9/S1/S8 © 2008 Tas¸an et al; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Individual researchers are struggling to keep up with the accelerating emergence of high-throughput biological data, and to extract information that relates to their specific questions. Integration of accumulated evidence should permit researchers to form fewer - and more accurate - hypotheses for further study through experimentation. Results: Here a method previously used to predict Gene Ontology (GO) terms for Saccharomyces cerevisiae (Tian et al.: Combining guilt-by-association and guilt-by-profiling to predict Saccharomyces cerevisiae gene function. Genome Biol 2008, 9(Suppl 1):S7) is applied to predict GO terms and phenotypes for 21,603 Mus musculus genes, using a diverse collection of integrated data sources (including expression, interaction, and sequence-based data). -

The Oncogenic Potential of BK-Polyomavirus Is Linked to Viral Integration Into the Human Genome
Journal of Pathology J Pathol 2015; 237: 379–389 ORIGINAL PAPER Published online 19 August 2015 in Wiley Online Library (wileyonlinelibrary.com) DOI: 10.1002/path.4584 The oncogenic potential of BK-polyomavirus is linked to viral integration into the human genome Daniel J Kenan,1 Piotr A Mieczkowski,2 Raquel Burger-Calderon,3 Harsharan K Singh1 and Volker Nickeleit1* 1 Division of Nephropathology, Department of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, NC, USA 2 Department of Genetics, University of North Carolina, Chapel Hill, NC, USA 3 School of Dentistry, University of North Carolina, Chapel Hill, NC, USA *Correspondence to: V Nickeleit, UNC Division of Nephropathology, University of North Carolina Medical School, Department of Pathology and Laboratory Medicine, Brinkhous-Bullitt Building, Room 409, Campus Box 7525, Chapel Hill, NC 27599–7525, USA. E-mail: [email protected] Abstract It has been suggested that BK-polyomavirus is linked to oncogenesis via high expression levels of large T-antigen in some urothelial neoplasms arising following kidney transplantation. However, a causal association between BK-polyomavirus, large T-antigen expression and oncogenesis has never been demonstrated in humans. Here we describe an investigation using high-throughput sequencing of tumour DNA obtained from an urothelial carcinoma arising in a renal allograft. We show that a novel BK-polyomavirus strain, named CH-1, is integrated into exon 26 of the myosin-binding protein C1 gene (MYBPC1) on chromosome 12 in tumour cells but not in normal renal cells. Integration of the BK-polyomavirus results in a number of discrete alterations in viral gene expression, including: (a) disruption of VP1 protein expression and robust expression of large T-antigen; (b) preclusion of viral replication; and (c) deletions in the non-coding control region (NCCR), with presumed alterations in promoter feedback loops. -

Pivotal Role of the Muscle-Contraction Pathway in Cryptorchidism And
Cannistraci et al. BMC Medical Genomics 2013, 6:5 http://www.biomedcentral.com/1755-8794/6/5 RESEARCH ARTICLE Open Access Pivotal role of the muscle-contraction pathway in cryptorchidism and evidence for genomic connections with cardiomyopathy pathways in RASopathies Carlo V Cannistraci1,2,3*†, Jernej Ogorevc4†, Minja Zorc4†, Timothy Ravasi1, Peter Dovc4 and Tanja Kunej4* Abstract Background: Cryptorchidism is the most frequent congenital disorder in male children; however the genetic causes of cryptorchidism remain poorly investigated. Comparative integratomics combined with systems biology approach was employed to elucidate genetic factors and molecular pathways underlying testis descent. Methods: Literature mining was performed to collect genomic loci associated with cryptorchidism in seven mammalian species. Information regarding the collected candidate genes was stored in MySQL relational database. Genomic view of the loci was presented using Flash GViewer web tool (http://gmod.org/wiki/Flashgviewer/). DAVID Bioinformatics Resources 6.7 was used for pathway enrichment analysis. Cytoscape plug-in PiNGO 1.11 was employed for protein-network-based prediction of novel candidate genes. Relevant protein-protein interactions were confirmed and visualized using the STRING database (version 9.0). Results: The developed cryptorchidism gene atlas includes 217 candidate loci (genes, regions involved in chromosomal mutations, and copy number variations) identified at the genomic, transcriptomic, and proteomic level. Human orthologs of the collected candidate loci were presented using a genomic map viewer. The cryptorchidism gene atlas is freely available online: http://www.integratomics-time.com/cryptorchidism/. Pathway analysis suggested the presence of twelve enriched pathways associated with the list of 179 literature-derived candidate genes. Additionally, a list of 43 network-predicted novel candidate genes was significantly associated with four enriched pathways. -

Four Parameters Increase the Sensitivity and Specificity of The
Journal of Human Genetics (2012) 57, 368–374 & 2012 The Japan Society of Human Genetics All rights reserved 1434-5161/12 $32.00 www.nature.com/jhg ORIGINAL ARTICLE Four parameters increase the sensitivity and specificity of the exon array analysis and disclose 25 novel aberrantly spliced exons in myotonic dystrophy This article has been corrected since advance Online Publication, and a corrigendum is also printed in this issue Yoshihiro Yamashita1,6, Tohru Matsuura1,2,6, Jun Shinmi1, Yoshinobu Amakusa1, Akio Masuda1, Mikako Ito1, Masanobu Kinoshita3, Hirokazu Furuya4, Koji Abe2, Tohru Ibi5, Ko Sahashi5 and Kinji Ohno1 Myotonic dystrophy type 1 (DM1) is an RNA gain-of-function disorder in which abnormally expanded CTG repeats of DMPK sequestrate a splicing trans-factor MBNL1 and upregulate another splicing trans-factor CUGBP1. To identify a diverse array of aberrantly spliced genes, we performed the exon array analysis of DM1 muscles. We analyzed 72 exons by RT-PCR and found that 27 were aberrantly spliced, whereas 45 were not. Among these, 25 were novel and especially splicing aberrations of LDB3 exon 4 and TTN exon 45 were unique to DM1. Retrospective analysis revealed that four parameters efficiently detect aberrantly spliced exons: (i) the signal intensity is high; (ii) the ratio of probe sets with reliable signal intensities (that is, detection above background P-value ¼ 0.000) is high within a gene; (iii) the splice index (SI) is high; and (iv) SI is deviated from SIs of the other exons that can be estimated by calculating the deviation value (DV). Application of the four parameters gave rise to a sensitivity of 77.8% and a specificity of 95.6% in our data set. -

Gene Ontology Analysis of Arthrogryposis (Multiple Congenital Contractures)
Received: 5 March 2019 Revised: 13 June 2019 Accepted: 17 July 2019 DOI: 10.1002/ajmg.c.31733 RESEARCH ARTICLE Gene ontology analysis of arthrogryposis (multiple congenital contractures) Jeff Kiefer1 | Judith G. Hall2,3 1Systems Oncology, Scottsdale, Arizona Abstract 2Department of Medical Genetics, University of British Columbia and BC Children's In 2016, we published an article applying Gene Ontology Analysis to the genes that had Hospital, Vancouver, British Columbia, Canada been reported to be associated with arthrogryposis (multiple congenital contractures) (Hall 3Department of Pediatrics, University of & Kiefer, 2016). At that time, 320 genes had been reported to have mutations associated British Columbia and BC Children's Hospital, Vancouver, British Columbia, Canada with arthrogryposis. All were associated with decreased fetal movement. These 320 genes were analyzed by biological process and cellular component categories, and yielded 22 Correspondence Judith G. Hall, Department of Medical distinct groupings. Since that time, another 82 additional genes have been reported, now Genetics, BC Children's Hospital, 4500 Oak totaling 402 genes, which when mutated, are associated with arthrogryposis (arthrogryposis Street, Room C234, Vancouver, British Columbia V6H 3N1, Canada. multiplex congenita). So, we decided to update the analysis in order to stimulate further Email: [email protected] research and possible treatment. Now, 29 groupings can be identified, but only 19 groups have more than one gene. KEYWORDS arthrogryposis, developmental pathways, enrichment analysis, gene ontology, multiple congenital contractures 1 | INTRODUCTION polyhydramnios, decreased gut mobility and shortened gut, short umbili- cal cord, skin changes, and multiple joints with limitation of movement, Arthrogryposis is the term that has been used for the last century including limbs, jaw, and spine).