SN2 Reaction Rate Enhancement by Β-Cyclodextrin at the Liquid/Liquid Interface
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bsc Chemistry
Subject Chemistry Paper No and Title 05, ORGANIC CHEMISTRY-II (REACTION MECHANISM-I) Module No and Title 15, The Neighbouring Mechanism, Neighbouring Group Participation by π and σ Bonds Module Tag CHE_P5_M15 CHEMISTRY PAPER :5, ORGANIC CHEMISTRY-II (REACTION MECHANISM-I) MODULE: 15 , The Neighbouring Mechanism, Neighbouring Group Participation by π and σ Bonds TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. NGP Participation 3.1 NGP by Heteroatom Lone Pairs 3.2 NGP by alkene 3.3 NGP by Cyclopropane, Cyclobutane or a Homoallyl group 3.4 NGP by an Aromatic Ring 4. Neighbouring Group Participation on SN2 Reactions 5. Neighbouring Group Participation on SN1 Reactions 6. Neighbouring Group and Rearrangement 7. Examples 8. Summary CHEMISTRY PAPER :5, ORGANIC CHEMISTRY-II (REACTION MECHANISM-I) MODULE: 15 , The Neighbouring Mechanism, Neighbouring Group Participation by π and σ Bonds 1. Learning Outcomes After studying this module, you shall be able to Know about NGP reaction Learn reaction mechanism of NGP reaction Identify stereochemistry of NGP reaction Evaluate the factors affecting the NGP reaction Analyse Phenonium ion, NGP by alkene, and NGP by heteroatom. 2. Introduction The reaction centre (carbenium centre) has direct interaction with a lone pair of electrons of an atom or with the electrons of s- or p-bond present within the parent molecule but these are not in conjugation with the reaction centre. A distinction is sometimes made between n, s and p- participation. An increase in rate due to Neighbouring Group Participation (NGP) is known as "anchimeric assistance". "Synartetic acceleration" happens to be the special case of anchimeric assistance and applies to participation by electrons binding a substituent to a carbon atom in a β- position relative to the leaving group attached to the α-carbon atom. -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

SN2 , SN1 , E2 , & E1: Substitution and Elimination Reactions
SN2 , SN1 , E2 , & E1: Substitution and Elimination Reactions • Nucleophilic Substitution Reactions (SN2 and SN1) replace a leaving group with a nucleophile (Nu: or Nu: - ) • Elimination Reactions (E2 and E1) generate a double bond by loss of " A+ " and " B: - " • They may compete with each other Nucleophilic Substitution Reactions - SN2 Reaction: • Reaction is: o Stereospecific (Walden Inversion of configuration) o Concerted - all bonds form and break at same time o Bimolecular - rate depends on concentration of both nucleophile and substrate • Substrate: o Best if primary (one substituent on carbon bearing leaving group) o works if secondary, fails if tertiary • Nucleophile: o Best if more reactive (i.e. more anionic or more basic) • Leaving Group: Best if more stable (i.e. can support negative charge well): o TsO- (very good) > I- > Br- > Cl- > F- (poor) o RF , ROH , ROR , RNH2 are NEVER Substrates for SN2 reactions o Leaving Groups on double-bonded carbons are never replaced by SN2 reactions • Solvent: Polar Aprotic (i.e. no OH) is best. o For example dimethylsulfoxide ( CH3SOCH3 ), dimethylformamide ( HCON(CH3)2 ), acetonitrile ( CH3CN ). o Protic solvents (e.g. H2O or ROH) deactivate nucleophile by hydrogen bonding but can be used in some case Intro Chem Handouts Substitution & Elimination Reactions Page 1 of 3 Nucleophilic Substitution Reactions – SN1 Reaction: • Reaction is: o Non-stereospecific (attack by nucleophile occurs from both sides) o Non-concerted - has carbocation intermediate o Unimolecular - rate depends on concentration of only the substrate • Substrate: o Best if tertiary or conjugated (benzylic or allylic) carbocation can be formed as leaving group departs o never primary • Nucleophile: o Best if more reactive (i.e. -

Elimination Reactions Are Described
Introduction In this module, different types of elimination reactions are described. From a practical standpoint, elimination reactions widely used for the generation of double and triple bonds in compounds from a saturated precursor molecule. The presence of a good leaving group is a prerequisite in most elimination reactions. Traditional classification of elimination reactions, in terms of the molecularity of the reaction is employed. How the changes in the nature of the substrate as well as reaction conditions affect the mechanism of elimination are subsequently discussed. The stereochemical requirements for elimination in a given substrate and its consequence in the product stereochemistry is emphasized. ELIMINATION REACTIONS Objective and Outline beta-eliminations E1, E2 and E1cB mechanisms Stereochemical considerations of these reactions Examples of E1, E2 and E1cB reactions Alpha eliminations and generation of carbene I. Basics Elimination reactions involve the loss of fragments or groups from a molecule to generate multiple bonds. A generalized equation is shown below for 1,2-elimination wherein the X and Y from two adjacent carbon atoms are removed, elimination C C C C -XY X Y Three major types of elimination reactions are: α-elimination: two atoms or groups are removed from the same atom. It is also known as 1,1-elimination. H R R C X C + HX R Both H and X are removed from carbon atom here R Carbene β-elimination: loss of atoms or groups on adjacent atoms. It is also H H known as 1,2- elimination. R C C R R HC CH R X H γ-elimination: loss of atoms or groups from the 1st and 3rd positions as shown below. -

Oct 16 2018 Chem 261 Notes
CHEM 261 October 9, 2018 RECALL: Identification of Chiral and Achiral (not chiral) compounds Example: Diaminopimelic acid - The above molecule is achiral even though there are stereogenic center (s), because there is symmetry within the molecule - These kinds of molecules are called meso compounds, which are compounds that contain stereocenters yet because of their symmetry, have mirror images that can be superimposed. - All achiral molecules, including meso compounds do not rotate polarized light (i.e. [α]D = 0) - Diaminopimelic acid - a component of bacterial cell wall and biosynthetic precursor to the amino acid known as lysine - this R,S diaminopimelic acid (above) is a diastereomer of the enantiomers (S,S or R,R diaminopimelic acid) below: Mirror O O O O C S C C R C HO S OH HO R OH NH2 NH2 NH2 NH2 chiral chiral Enantiomers A racemic mixture (racemate) of two enantiomers in a 1:1 ratio also has an [α]D = 0 Resolution of Enantiomers Definition: separation of two enantiomers - Requires a chiral reagent The starting material lactic acids are enantiomers of each other. By reacting enantiomers to make a salt with an enantiomer of MDMA (another chiral molecule), one can obtain salts which are now diastereomers of each other (RS and RR). The resulting diastereomers have different melting points, boiling points, solubilities, and can be separated by crystallization. >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>> Substitution Reactions Remember: Radical Substitution light R-H + X2 R-X + H-X Proceeds by a radical mechanism Alkane X=F, Cl, Br Alkyl Halide Not I Ionic Substitution: M Nu: + R-X Nu-R + :X M Nucleophile Alkyl Halide (Seeks positive charge) Nucleophile is a subtsnace that seeks positive charge Types of Nucleophilic Substitution (SN) SN1 - rate depends on 1 concentration SN2 - The rate is dependent on the concentration of the nucleophile and the nucleophile (2 concentrations) Sn2 Mechanism Reverse reaction will not occur. -

Concerted Nucleophilic Aromatic Substitution Reactions Simon Rohrbach+, Andrew J
Angewandte Reviews Chemie International Edition: DOI: 10.1002/anie.201902216 Nucleophilic Aromatic Substitution German Edition: DOI: 10.1002/ange.201902216 Concerted Nucleophilic Aromatic Substitution Reactions Simon Rohrbach+, Andrew J. Smith+, Jia Hao Pang+, Darren L. Poole, Tell Tuttle,* Shunsuke Chiba,* and John A. Murphy* Keywords: Dedicated to Professor Koichi concerted reactions Narasaka on the occasion of ·cSNAr mechanism · his 75th birthday Meisenheimer complex · nucleophilicaromatic substitution Angewandte Chemie &&&& 2019 The Authors. Published by Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2019, 58,2–23 Ü Ü These are not the final page numbers! Angewandte Reviews Chemie Recent developments in experimental and computational From the Contents chemistry have identified a rapidly growing class of nucleophilic 1. Aromatic Substitution Reactions 3 aromatic substitutions that proceed by concerted (cSNAr) rather than classical, two-step, SNAr mechanisms. Whereas traditional 2. Some Contributions by SNAr reactions require substantial activation of the aromatic ring Computational Studies 6 by electron-withdrawing substituents, such activating groups are not mandatory in the concerted pathways. 3. Fluorodeoxygenation of Phenols and Derivatives 9 4. Aminodeoxygenation of Phenol 1. Aromatic Substitution Reactions Derivatives 10 Substitution reactions on aromatic rings are central to 5. Hydrides as Nucleophiles 11 organic chemistry. Besides the commonly encountered elec- trophilic aromatic substitution,[1] other mechanisms include 6. P, N, Si, C Nucleophiles 13 [2,3] SNAr nucleophilic aromatic substitutions and the distinct [4] but related SNArH and vicarious nucleophilic substitutions, 7. Organic Rearrangements via Spiro substitutions brought about through benzyne intermedi- Species: Intermediates or Transition ates,[5,6] radical mechanisms including electron transfer- States? 14 [7] based SRN1 reactions and base-promoted homolytic aro- matic substitution (BHAS) couplings,[8] sigmatropic rear- 8. -

Transition-Metal Catalysis of Nucleophilic Substitution Reactions: a Radical Alternative to SN1 and SN2 Processes Gregory C
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. Outlook http://pubs.acs.org/journal/acscii Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes Gregory C. Fu* Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States ABSTRACT: Classical methods for achieving nucleophilic substitutions of alkyl electrophiles (SN1 and SN2) have limited scope and are not generally amenable to enantioselective variants that employ readily available racemic electrophiles. Radical-based pathways catalyzed by chiral transition-metal complexes provide an attractive approach to addressing these limitations. ■ UNSOLVED CHALLENGES IN NUCLEOPHILIC SUBSTITUTION REACTIONS OF ALKYL ELECTROPHILES Nucleophilic substitution of an alkyl electrophile is an extremely useful strategy in organic synthesis (Figure 1). SN1 and SN2 reactions are the two classical pathways for achieving this process; both have significant limitations.1,2 Figure 2. Limitations of the SN1 reaction. nucleophile and the electrophile, and it is therefore often unsuccessful in the case of hindered primary (e.g., neopentyl) Figure 1. Nucleophilic substitution reactions of alkyl electrophiles: and secondary electrophiles (Figure 3). Furthermore, SN2 SN1 and SN2 reactions. reactions are generally conducted under Brønsted-basic conditions, which can -
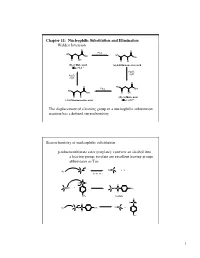
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion O O PCl5 HO HO OH OH O OH O Cl (S)-(-) Malic acid (+)-2-Chlorosuccinic acid [a]D= -2.3 ° Ag2O, H2O Ag2O, H2O O O HO PCl5 OH HO OH O OH O Cl (R)-(+) Malic acid (-)-2-Chlorosuccinic acid [a]D= +2.3 ° The displacement of a leaving group in a nucleophilic substitution reaction has a defined stereochemistry Stereochemistry of nucleophilic substitution p-toluenesulfonate ester (tosylate): converts an alcohol into a leaving group; tosylate are excellent leaving groups. abbreviates as Tos C X Nu C + X- Nu: X= Cl, Br, I O Cl S O O + C OH C O S CH3 O CH3 tosylate O -O S O O Nu C + Nu: C O S CH3 O CH3 1 O Tos-Cl - H3C O O H + TosO - H O H pyridine H O Tos O CH3 [a]D= +33.0 [a]D= +31.1 [a]D= -7.06 HO- HO- O - Tos-Cl H3C O - H O O TosO + H O H pyridine Tos H O CH3 [a]D= -7.0 [a]D= -31.0 [a]D= -33.2 The nucleophilic substitution reaction “inverts” the Stereochemistry of the carbon (electrophile)- Walden inversion Kinetics of nucleophilic substitution Reaction rate: how fast (or slow) reactants are converted into product (kinetics) Reaction rates are dependent upon the concentration of the reactants. (reactions rely on molecular collisions) H H Consider: HO C _ _ C Br Br HO H H H H At a given temperature: If [OH-] is doubled, then the reaction rate may be doubled If [CH3-Br] is doubled, then the reaction rate may be doubled A linear dependence of rate on the concentration of two reactants is called a second-order reaction (molecularity) 2 H H HO C _ _ C Br Br HO H H H H Reaction rates (kinetic) can be expressed mathematically: reaction rate = disappearance of reactants (or appearance of products) For the disappearance of reactants: - rate = k [CH3Br] [OH ] [CH3Br] = CH3Br concentration [OH-] = OH- concentration k= constant (rate constant) L mol•sec For the reaction above, product formation involves a collision between both reactants, thus the rate of the reaction is dependent upon the concentration of both. -

PRACTICE EXERCISE Sn1 and Sn2 Reactions Δ
ORGANIC CHEMISTRY I – PRACTICE EXERCISE Sn1 and Sn2 Reactions 1) Which of the following best represents the carbon-chlorine bond of methyl chloride? H H H H H - - + d+ d d d d+ d+ d- d- C Cl C Cl C Cl C Cl C Cl H H H H H H H H H H I II III IV V 2) Provide a detailed, stepwise mechanism for the reaction below. Br + CN CN + Br 3) Rank the species below in order of increasing nucleophilicity in hydroxylic solvents: CH3CO2- CH3S- HO- H2O 4) Give a stereochemical structure of the product from the reaction between (S)-2-iodopentane and KCN in DMF (dimethyl formamide, a good polar solvent for ionic reagents). 5) Consider the reaction of (CH3)3CO- with iodomethane. Will the reaction rate increase, decrease, or remain the same if the concentration of iodomethane is increased? Explain. 6) Which of the following compounds will undergo an Sn2 reaction most readily? A) (CH3)3CCH2I B) (CH3)3CCl C) (CH3)2CHI D) (CH3)2CHCH2CH2CH2I E) (CH3)2CHCH2CH2CH2Cl 7) What is the major organic product in the following reaction? CH3S Br CH3 acetone 8) Would 2-chloropropane or 1-chloro-2,2-dimethylpropane undergo substitution faster with Na+ -CCH? Give the structure of the substitution product. 9) t-butyl chloride undergoes solvolysis in 70% water/30% acetone at a rate slower than in 80% water/20% acetone. Explain. 10) Provide the major organic product of the reaction below and a detailed, stepwise mechanism which accounts for its formation. Br CH3OH D CH2CH3 11) Sn2 reactions involving chiral electrophiles usually proceed with: A) inversion of configuration B) slightly more inversion than retention. -

The Sn2 Reaction: a Theoretical-Computational Analysis of a Simple and Very Interesting Mechanism †
Proceedings The Sn2 Reaction: A Theoretical-Computational Analysis of a Simple and Very Interesting † Mechanism Matías Capurso ‡, Rodrigo Gette ‡, Gabriel Radivoy and Viviana Dorn * Instituto de Química del Sur (INQUISUR-CONICET), Depto. de Química, Universidad Nacional del Sur, Av. Alem 1253, B8000CPB Bahía Blanca, Argentina; [email protected] (M.C.); [email protected] (R.G.); [email protected] (G.R.) * Correspondence: [email protected] † Presented at the 23rd International Electronic Conference on Synthetic Organic Chemistry, 15 November–15 December 2019; Available online: https://ecsoc-23.sciforum.net/. ‡ Both authors contributed equally to this manuscript. Published: 14 November 2019 Abstract: Bimolecular nucleophilic substitution (SN2) reaction is one of the most frequently processes chosen as model mechanism to introduce undergraduate chemistry students to computational chemistry methodology. In this work, we performed a computational analysis for the ionic SN2 reaction, where the nucleophile charged (X−; X=F, Cl, Br, I) attacks the carbon atom of the substrate (CH3Cl) through a backside pathway, and simultaneously, the leaving group is displaced (Cl−). The calculations were performed applying DFT methods with the Gaussian09 program, the B3LYP functional, the 6-31+G* basis set for all atoms except iodine (6-311G*), and the solvents effects (acetonitrile and cyclohexane) were evaluated with the PCM model. We evaluated the potential energy surface (PES) for the mentioned reaction considering the reactants, the formation of an initial complex between the nucleophile and the substrate, the transition state, a final complex where the leaving group is still bound to the substrate and the products. We analyzed the atomic charge (ESP) and the bond distance throughout the process. -

Chem 51C Chapter 22 Notes
Lecture Notes Chem 51C S. King Chapter 22 Carboxylic Acids and their Derivatives: Nucleophilic Acyl Substitution I. Structure and Physical Properties: Type 2 carbonyl compounds (carboxylic acids and derivatives) contain the carbonyl group bonded to an atom that has at least one pair of non-bonding electrons. O O C L = Cl , NH2 , OCH3 , OH , OCR R L Physical properties: O O O O O O O CH3 OH CH3 Cl CH3 OEt CH3 NH2 CH3 O CH3 CH3(CH2)8 OH BP Sol. in H2O • Both carboxylic acids and 1° and 2° amides can form hydrogen bonds, and therefore have higher bp’s and greater solubility that other Type 2 compounds. • Amides have higher boiling points than carboxylic acids because they have very strong dipole-dipole attractions (the resonance contributor with separated charges contributes significantly to the overall structure of the molecule.) O O H NH2 H OH • Tertiary amides have significantly lower boiling points despite the fact that they have higher molecular weights because they cannot form hydrogen bonds. O O CH3 NH2 CH3 N(CH3)2 53 II. Some Famous Type 2 Carbonyl Compounds (Carboxylic Acid Derivatives): H CH3 NH2 N CO2CH3 O N O2CPh O Novocaine H cocaine CH3 O O N N NH Cl N N Ph CH3 LSD Diazepam (Valium) O H3C O H H CH3 N N CH2CNH S CH3 N N N O CH3 O CH – + caffeine 3 Penicillin G COO K O O CH2CH3 CO2H HN Ph O N O H H OH Phenobarbital prostaglandin A2 O CO2H CH3COC H2 O CH C 3 HO CH CH CO H 2 2 2 2 benzyl acetate CH COCH CH CHCH OH 3 2 2 3 isopentyl acetate citric acid 54 III. -

Synopsis of SN1, SN2, E1 and E2 Reactions
Synopsis of SN1, SN2, E1 and E2 Reactions Subsitution Nucleophilic Unimolecular (SN1) Subsitution Nucleophilic Bimolecular (SN2) Rate = k [RX] Rate = k [RX] [Nuc] Kinetics Only substrate (RX) involved in rate- Rate-determining step involves both substrate and determining step (slow step). nucleophile Weak nucleophiles (i.e. neutral); on periodic Strong nucleophile - negatively charged better (eg. Nucleophile table: nucleophile increases strength going to methoxide better than methanol). the left and going down Relatively uninhindered alkyl halide; methyl>1˚>2˚. Substrates that form relatively stable Due to steric hinderence, 3˚ virtually unreactive by S 2 Substrate Reactivity carbocations; 3˚>2˚>>1˚>methyl N (in the presence of good nucleophile/base it undergoes (1˚ & methyl virtually unreactive by S 1) N E2) Stereochemistry Results in racemization. always inversion of configuration Occurs if more stable carbocation can form (eg. Rearrangements Do not occur 2˚ to 3˚) Solvents that leave nucleophile (anion) relatively Highly ionizing solvents; polar, protic favored unencumbered. Theoretically, apolar solvents are the Solvents (eg. water, methanol, ethanol, alcohol, best but, in practice, a polar aprotic (eg. DMF, DMSO, ammonia) THF) is necessary to help dissolve the nucleophile. Occurs in several steps, the rate-determining One-step mechanism; no intermediates; transition state Reaction Mechanism step is the unimolecular ionization of the involves both nucleophile and substrate (bimolecular) substrate to form a carbocation intermediate In the TS, R takes on δ+ (leading to Nucleophile displaces leaving group. Both incoming Transition State (TS) carbocation) and X takes on δ- (leading to nucleophile (Nuc-) and exiting leaving group (X-) bear a leaving group) δ- charge in the TS.