Multiple Testing Procedures for Complex Structured Hypotheses and Directional Decisions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

05 36534Nys130620 31
Monte Carlos study on Power Rates of Some Heteroscedasticity detection Methods in Linear Regression Model with multicollinearity problem O.O. Alabi, Kayode Ayinde, O. E. Babalola, and H.A. Bello Department of Statistics, Federal University of Technology, P.M.B. 704, Akure, Ondo State, Nigeria Corresponding Author: O. O. Alabi, [email protected] Abstract: This paper examined the power rate exhibit by some heteroscedasticity detection methods in a linear regression model with multicollinearity problem. Violation of unequal error variance assumption in any linear regression model leads to the problem of heteroscedasticity, while violation of the assumption of non linear dependency between the exogenous variables leads to multicollinearity problem. Whenever these two problems exist one would faced with estimation and hypothesis problem. in order to overcome these hurdles, one needs to determine the best method of heteroscedasticity detection in other to avoid taking a wrong decision under hypothesis testing. This then leads us to the way and manner to determine the best heteroscedasticity detection method in a linear regression model with multicollinearity problem via power rate. In practices, variance of error terms are unequal and unknown in nature, but there is need to determine the presence or absence of this problem that do exist in unknown error term as a preliminary diagnosis on the set of data we are to analyze or perform hypothesis testing on. Although, there are several forms of heteroscedasticity and several detection methods of heteroscedasticity, but for any researcher to arrive at a reasonable and correct decision, best and consistent performed methods of heteroscedasticity detection under any forms or structured of heteroscedasticity must be determined. -

The Effects of Simplifying Assumptions in Power Analysis
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Public Access Theses and Dissertations from Education and Human Sciences, College of the College of Education and Human Sciences (CEHS) 4-2011 The Effects of Simplifying Assumptions in Power Analysis Kevin A. Kupzyk University of Nebraska-Lincoln, [email protected] Follow this and additional works at: https://digitalcommons.unl.edu/cehsdiss Part of the Educational Psychology Commons Kupzyk, Kevin A., "The Effects of Simplifying Assumptions in Power Analysis" (2011). Public Access Theses and Dissertations from the College of Education and Human Sciences. 106. https://digitalcommons.unl.edu/cehsdiss/106 This Article is brought to you for free and open access by the Education and Human Sciences, College of (CEHS) at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Public Access Theses and Dissertations from the College of Education and Human Sciences by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Kupzyk - i THE EFFECTS OF SIMPLIFYING ASSUMPTIONS IN POWER ANALYSIS by Kevin A. Kupzyk A DISSERTATION Presented to the Faculty of The Graduate College at the University of Nebraska In Partial Fulfillment of Requirements For the Degree of Doctor of Philosophy Major: Psychological Studies in Education Under the Supervision of Professor James A. Bovaird Lincoln, Nebraska April, 2011 Kupzyk - i THE EFFECTS OF SIMPLIFYING ASSUMPTIONS IN POWER ANALYSIS Kevin A. Kupzyk, Ph.D. University of Nebraska, 2011 Adviser: James A. Bovaird In experimental research, planning studies that have sufficient probability of detecting important effects is critical. Carrying out an experiment with an inadequate sample size may result in the inability to observe the effect of interest, wasting the resources spent on an experiment. -

Introduction to Hypothesis Testing
Introduction to Hypothesis Testing OPRE 6301 Motivation . The purpose of hypothesis testing is to determine whether there is enough statistical evidence in favor of a certain belief, or hypothesis, about a parameter. Examples: Is there statistical evidence, from a random sample of potential customers, to support the hypothesis that more than 10% of the potential customers will pur- chase a new product? Is a new drug effective in curing a certain disease? A sample of patients is randomly selected. Half of them are given the drug while the other half are given a placebo. The conditions of the patients are then mea- sured and compared. These questions/hypotheses are similar in spirit to the discrimination example studied earlier. Below, we pro- vide a basic introduction to hypothesis testing. 1 Criminal Trials . The basic concepts in hypothesis testing are actually quite analogous to those in a criminal trial. Consider a person on trial for a “criminal” offense in the United States. Under the US system a jury (or sometimes just the judge) must decide if the person is innocent or guilty while in fact the person may be innocent or guilty. These combinations are summarized in the table below. Person is: Innocent Guilty Jury Says: Innocent No Error Error Guilty Error No Error Notice that there are two types of errors. Are both of these errors equally important? Or, is it as bad to decide that a guilty person is innocent and let them go free as it is to decide an innocent person is guilty and punish them for the crime? Or, is a jury supposed to be totally objective, not assuming that the person is either innocent or guilty and make their decision based on the weight of the evidence one way or another? 2 In a criminal trial, there actually is a favored assump- tion, an initial bias if you will. -

Power of a Statistical Test
Power of a Statistical Test By Smita Skrivanek, Principal Statistician, MoreSteam.com LLC What is the power of a test? The power of a statistical test gives the likelihood of rejecting the null hypothesis when the null hypothesis is false. Just as the significance level (alpha) of a test gives the probability that the null hypothesis will be rejected when it is actually true (a wrong decision), power quantifies the chance that the null hypothesis will be rejected when it is actually false (a correct decision). Thus, power is the ability of a test to correctly reject the null hypothesis. Why is it important? Although you can conduct a hypothesis test without it, calculating the power of a test beforehand will help you ensure that the sample size is large enough for the purpose of the test. Otherwise, the test may be inconclusive, leading to wasted resources. On rare occasions the power may be calculated after the test is performed, but this is not recommended except to determine an adequate sample size for a follow-up study (if a test failed to detect an effect, it was obviously underpowered – nothing new can be learned by calculating the power at this stage). How is it calculated? As an example, consider testing whether the average time per week spent watching TV is 4 hours versus the alternative that it is greater than 4 hours. We will calculate the power of the test for a specific value under the alternative hypothesis, say, 7 hours: The Null Hypothesis is H0: μ = 4 hours The Alternative Hypothesis is H1: μ = 6 hours Where μ = the average time per week spent watching TV. -

Confidence Intervals and Hypothesis Tests
Chapter 2 Confidence intervals and hypothesis tests This chapter focuses on how to draw conclusions about populations from sample data. We'll start by looking at binary data (e.g., polling), and learn how to estimate the true ratio of 1s and 0s with confidence intervals, and then test whether that ratio is significantly different from some baseline value using hypothesis testing. Then, we'll extend what we've learned to continuous measurements. 2.1 Binomial data Suppose we're conducting a yes/no survey of a few randomly sampled people 1, and we want to use the results of our survey to determine the answers for the overall population. 2.1.1 The estimator The obvious first choice is just the fraction of people who said yes. Formally, suppose we have samples x1,..., xn that can each be 0 or 1, and the probability that each xi is 1 is p (in frequentist style, we'll assume p is fixed but unknown: this is what we're interested in finding). We'll assume our samples are indendepent and identically distributed (i.i.d.), meaning that each one has no dependence on any of the others, and they all have the same probability p of being 1. Then our estimate for p, which we'll callp ^, or \p-hat" would be n 1 X p^ = x : n i i=1 Notice thatp ^ is a random quantity, since it depends on the random quantities xi. In statistical lingo,p ^ is known as an estimator for p. Also notice that except for the factor of 1=n in front, p^ is almost a binomial random variable (in particular, (np^) ∼ B(n; p)). -

Understanding Statistical Hypothesis Testing: the Logic of Statistical Inference
Review Understanding Statistical Hypothesis Testing: The Logic of Statistical Inference Frank Emmert-Streib 1,2,* and Matthias Dehmer 3,4,5 1 Predictive Society and Data Analytics Lab, Faculty of Information Technology and Communication Sciences, Tampere University, 33100 Tampere, Finland 2 Institute of Biosciences and Medical Technology, Tampere University, 33520 Tampere, Finland 3 Institute for Intelligent Production, Faculty for Management, University of Applied Sciences Upper Austria, Steyr Campus, 4040 Steyr, Austria 4 Department of Mechatronics and Biomedical Computer Science, University for Health Sciences, Medical Informatics and Technology (UMIT), 6060 Hall, Tyrol, Austria 5 College of Computer and Control Engineering, Nankai University, Tianjin 300000, China * Correspondence: [email protected]; Tel.: +358-50-301-5353 Received: 27 July 2019; Accepted: 9 August 2019; Published: 12 August 2019 Abstract: Statistical hypothesis testing is among the most misunderstood quantitative analysis methods from data science. Despite its seeming simplicity, it has complex interdependencies between its procedural components. In this paper, we discuss the underlying logic behind statistical hypothesis testing, the formal meaning of its components and their connections. Our presentation is applicable to all statistical hypothesis tests as generic backbone and, hence, useful across all application domains in data science and artificial intelligence. Keywords: hypothesis testing; machine learning; statistics; data science; statistical inference 1. Introduction We are living in an era that is characterized by the availability of big data. In order to emphasize the importance of this, data have been called the ‘oil of the 21st Century’ [1]. However, for dealing with the challenges posed by such data, advanced analysis methods are needed. -

Post Hoc Power: Tables and Commentary
Post Hoc Power: Tables and Commentary Russell V. Lenth July, 2007 The University of Iowa Department of Statistics and Actuarial Science Technical Report No. 378 Abstract Post hoc power is the retrospective power of an observed effect based on the sample size and parameter estimates derived from a given data set. Many scientists recommend using post hoc power as a follow-up analysis, especially if a finding is nonsignificant. This article presents tables of post hoc power for common t and F tests. These tables make it explicitly clear that for a given significance level, post hoc power depends only on the P value and the degrees of freedom. It is hoped that this article will lead to greater understanding of what post hoc power is—and is not. We also present a “grand unified formula” for post hoc power based on a reformulation of the problem, and a discussion of alternative views. Key words: Post hoc power, Observed power, P value, Grand unified formula 1 Introduction Power analysis has received an increasing amount of attention in the social-science literature (e.g., Cohen, 1988; Bausell and Li, 2002; Murphy and Myors, 2004). Used prospectively, it is used to determine an adequate sample size for a planned study (see, for example, Kraemer and Thiemann, 1987); for a stated effect size and significance level for a statistical test, one finds the sample size for which the power of the test will achieve a specified value. Many studies are not planned with such a prospective power calculation, however; and there is substantial evidence (e.g., Mone et al., 1996; Maxwell, 2004) that many published studies in the social sciences are under-powered. -
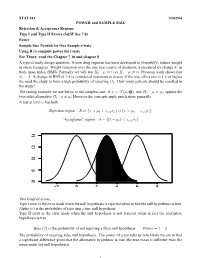
STAT 141 11/02/04 POWER and SAMPLE SIZE Rejection & Acceptance Regions Type I and Type II Errors (S&W Sec 7.8) Power
STAT 141 11/02/04 POWER and SAMPLE SIZE Rejection & Acceptance Regions Type I and Type II Errors (S&W Sec 7.8) Power Sample Size Needed for One Sample z-tests. Using R to compute power for t.tests For Thurs: read the Chapter 7.10 and chapter 8 A typical study design question: A new drug regimen has been developed to (hopefully) reduce weight in obese teenagers. Weight reduction over the one year course of treatment is measured by change X in body mass index (BMI). Formally we will test H0 : µ = 0 vs H1 : µ 6= 0. Previous work shows that σx = 2. A change in BMI of 1.5 is considered important to detect (if the true effect size is 1.5 or higher we need the study to have a high probability of rejecting H0. How many patients should be enrolled in the study? σ2 The testing example we use below is the simplest one: if x¯ ∼ N(µ, n ), test H0 : µ = µ0 against the two-sided alternative H1 : µ 6= µ0 However the concepts apply much more generally. A test at level α has both: Rejection region : R = {x¯ > µ0 + zα/2σx¯} ∪ {x¯ < µ0 − zα/2σx¯} “Acceptance” region : A = {|x¯ − µ0| < zα/2σx¯} 0.4 0.4 0.3 0.3 0.2 0.2 0.1 0.1 0.0 0.0 −2 0 2 4 6 8 Two kinds of errors: Type I error is the error made when the null hypothesis is rejected when in fact the null hypothesis is true. -

A Test of Independence in Two-Way Contingency Tables Based on Maximal Correlation
A TEST OF INDEPENDENCE IN TWO-WAY CONTINGENCY TABLES BASED ON MAXIMAL CORRELATION Deniz C. YenigÄun A Dissertation Submitted to the Graduate College of Bowling Green State University in partial ful¯llment of the requirements for the degree of DOCTOR OF PHILOSOPHY August 2007 Committee: G¶abor Sz¶ekely, Advisor Maria L. Rizzo, Co-Advisor Louisa Ha, Graduate Faculty Representative James Albert Craig L. Zirbel ii ABSTRACT G¶abor Sz¶ekely, Advisor Maximal correlation has several desirable properties as a measure of dependence, includ- ing the fact that it vanishes if and only if the variables are independent. Except for a few special cases, it is hard to evaluate maximal correlation explicitly. In this dissertation, we focus on two-dimensional contingency tables and discuss a procedure for estimating maxi- mal correlation, which we use for constructing a test of independence. For large samples, we present the asymptotic null distribution of the test statistic. For small samples or tables with sparseness, we use exact inferential methods, where we employ maximal correlation as the ordering criterion. We compare the maximal correlation test with other tests of independence by Monte Carlo simulations. When the underlying continuous variables are dependent but uncorre- lated, we point out some cases for which the new test is more powerful. iii ACKNOWLEDGEMENTS I would like to express my sincere appreciation to my advisor, G¶abor Sz¶ekely, and my co-advisor, Maria Rizzo, for their advice and help throughout this research. I thank to all the members of my committee, Craig Zirbel, Jim Albert, and Louisa Ha, for their time and advice. -

Statistical Power and P-Values: an Epistemic Interpretation Without Power Approach Paradoxes
Statistical Power and P-values: An Epistemic Interpretation Without Power Approach Paradoxes Guillaume Rochefort-Maranda December 16, 2017 Contents 1 Introduction 2 2 The Paradox 3 2.1 Technical Background . 3 2.2 Epistemic Interpretations . 4 2.3 A Paradox . 6 3 The Consensus 8 4 The Solution 10 5 Conclusion 13 1 1 Introduction It has been claimed that if statistical power and p-values are both used to measure the strength of our evidence for the null-hypothesis when the re- sults of our tests are not significant, then they can also be used to derive inconsistent epistemic judgements as we compare two different experi- ments. Those problematic derivations are known as power approach para- doxes. The consensus is that we can avoid them if we abandon the idea that statistical power can measure the strength of our evidence (Hoenig and Heisey 2001; Machery 2012). In this paper however, I put forward a different solution. I argue that every power approach paradox rests on an equivocation on ”strong evidence”. The main idea is that we need to make a careful distinction between (i) the evidence provided by the quality of the test and (ii) the evidence pro- vided by the outcome of the test. Both provide different types of evidence and their respective strength are to be evaluated differently. Without loss of generality1, I analyse only one power approach para- dox in order to reach this conclusion. But first, I set-up the frequentist framework within which we can find such a paradox. 1My analysis is without loss of generality because every other formulation of the para- dox rests on the same idea that I reject : power and p-values measure the same thing. -

The Probability of Not Committing a Type II Error Is Called the Power of a Hypothesis Test
The probability of not committing a Type II Error is called the Power of a hypothesis test. Effect Size To compute the power of the test, one offers an alternative view about the "true" value of the population parameter, assuming that the null hypothesis is false. The effect size is the difference between the true value and the value specified in the null hypothesis. Effect size = True value - Hypothesized value For example, suppose the null hypothesis states that a population mean is equal to 100. A researcher might ask: What is the probability of rejecting the null hypothesis if the true population mean is equal to 90? In this example, the effect size would be 90 - 100, which equals -10. Factors That Affect Power The power of a hypothesis test is affected by three factors. • Sample size ( n). Other things being equal, the greater the sample size, the greater the power of the test. • Significance level ( α). The higher the significance level, the higher the power of the test. If you increase the significance level, you reduce the region of acceptance. As a result, you are more likely to reject the null hypothesis. This means you are less likely to accept the null hypothesis when it is false; i.e., less likely to make a Type II error. Hence, the power of the test is increased. • The "true" value of the parameter being tested. The greater the difference between the "true" value of a parameter and the value specified in the null hypothesis, the greater the power of the test. -

Simulation-Based Power-Analysis for Factorial ANOVA Designs Daniel Lakens1 & Aaron R
Simulation-Based Power-Analysis for Factorial ANOVA Designs Daniel Lakens1 & Aaron R. Caldwell2,3 1 Human-Technology Interaction Group, Eindhoven University of Technology, The Netherlands 2 Department of Health, Human Performance and Recreation, University of Arkansas, USA 3 Thermal and Mountain Medicine Division, U.S. Army Research Institute of Environmental Medicine, USA Researchers often rely on analysis of variance (ANOVA) when they report results of experi- ments. To ensure a study is adequately powered to yield informative results when performing an ANOVA, researchers can perform an a-priori power analysis. However, power analysis for factorial ANOVA designs is often a challenge. Current software solutions do not allow power analyses for complex designs with several within-subject factors. Moreover, power analyses often need partial eta-squared or Cohen’s f as input, but these effect sizes are not intuitive and do not generalize to different experimental designs. We have created the R package Superpower and online Shiny apps to enable researchers without extensive programming experience to perform simulation-based power analysis for ANOVA designs of up to three within- or between-subject factors. Predicted effects are entered by specifying means, standard deviations, and for within- subject factors the correlations. The simulation provides the statistical power for all ANOVA main effects, interactions, and individual comparisons. The software can plot power across a range of sample sizes, can control for multiple comparisons, and can compute power when the homogeneity or sphericity assumptions are violated. This tutorial will demonstrate how to perform a-priori power analysis to design informative studies for main effects, interactions, and individual comparisons, and highlights important factors that determine the statistical power for factorial ANOVA designs.