A Novel PAX5 Rearrangement in TCF3
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Integrative Clinical Sequencing in the Management of Refractory Or
Supplementary Online Content Mody RJ, Wu Y-M, Lonigro RJ, et al. Integrative Clinical Sequencing in the Management of Children and Young Adults With Refractory or Relapsed CancerJAMA. doi:10.1001/jama.2015.10080. eAppendix. Supplementary appendix This supplementary material has been provided by the authors to give readers additional information about their work. © 2015 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/29/2021 SUPPLEMENTARY APPENDIX Use of Integrative Clinical Sequencing in the Management of Pediatric Cancer Patients *#Rajen J. Mody, M.B.B.S, M.S., *Yi-Mi Wu, Ph.D., Robert J. Lonigro, M.S., Xuhong Cao, M.S., Sameek Roychowdhury, M.D., Ph.D., Pankaj Vats, M.S., Kevin M. Frank, M.S., John R. Prensner, M.D., Ph.D., Irfan Asangani, Ph.D., Nallasivam Palanisamy Ph.D. , Raja M. Rabah, M.D., Jonathan R. Dillman, M.D., Laxmi Priya Kunju, M.D., Jessica Everett, M.S., Victoria M. Raymond, M.S., Yu Ning, M.S., Fengyun Su, Ph.D., Rui Wang, M.S., Elena M. Stoffel, M.D., Jeffrey W. Innis, M.D., Ph.D., J. Scott Roberts, Ph.D., Patricia L. Robertson, M.D., Gregory Yanik, M.D., Aghiad Chamdin, M.D., James A. Connelly, M.D., Sung Choi, M.D., Andrew C. Harris, M.D., Carrie Kitko, M.D., Rama Jasty Rao, M.D., John E. Levine, M.D., Valerie P. Castle, M.D., Raymond J. Hutchinson, M.D., Moshe Talpaz, M.D., ^Dan R. Robinson, Ph.D., and ^#Arul M. Chinnaiyan, M.D., Ph.D. CORRESPONDING AUTHOR (S): # Arul M. -

Supplementary Table 1 Double Treatment Vs Single Treatment
Supplementary table 1 Double treatment vs single treatment Probe ID Symbol Gene name P value Fold change TC0500007292.hg.1 NIM1K NIM1 serine/threonine protein kinase 1.05E-04 5.02 HTA2-neg-47424007_st NA NA 3.44E-03 4.11 HTA2-pos-3475282_st NA NA 3.30E-03 3.24 TC0X00007013.hg.1 MPC1L mitochondrial pyruvate carrier 1-like 5.22E-03 3.21 TC0200010447.hg.1 CASP8 caspase 8, apoptosis-related cysteine peptidase 3.54E-03 2.46 TC0400008390.hg.1 LRIT3 leucine-rich repeat, immunoglobulin-like and transmembrane domains 3 1.86E-03 2.41 TC1700011905.hg.1 DNAH17 dynein, axonemal, heavy chain 17 1.81E-04 2.40 TC0600012064.hg.1 GCM1 glial cells missing homolog 1 (Drosophila) 2.81E-03 2.39 TC0100015789.hg.1 POGZ Transcript Identified by AceView, Entrez Gene ID(s) 23126 3.64E-04 2.38 TC1300010039.hg.1 NEK5 NIMA-related kinase 5 3.39E-03 2.36 TC0900008222.hg.1 STX17 syntaxin 17 1.08E-03 2.29 TC1700012355.hg.1 KRBA2 KRAB-A domain containing 2 5.98E-03 2.28 HTA2-neg-47424044_st NA NA 5.94E-03 2.24 HTA2-neg-47424360_st NA NA 2.12E-03 2.22 TC0800010802.hg.1 C8orf89 chromosome 8 open reading frame 89 6.51E-04 2.20 TC1500010745.hg.1 POLR2M polymerase (RNA) II (DNA directed) polypeptide M 5.19E-03 2.20 TC1500007409.hg.1 GCNT3 glucosaminyl (N-acetyl) transferase 3, mucin type 6.48E-03 2.17 TC2200007132.hg.1 RFPL3 ret finger protein-like 3 5.91E-05 2.17 HTA2-neg-47424024_st NA NA 2.45E-03 2.16 TC0200010474.hg.1 KIAA2012 KIAA2012 5.20E-03 2.16 TC1100007216.hg.1 PRRG4 proline rich Gla (G-carboxyglutamic acid) 4 (transmembrane) 7.43E-03 2.15 TC0400012977.hg.1 SH3D19 -

Characterization of SPECC1L Function in Cell Adhesion and Migration in Craniofacial Morphogenesis by © 2017 Nathan Wilson B.Sc., Rockhurst University, 2010
Characterization of SPECC1L function in cell adhesion and migration in craniofacial morphogenesis By © 2017 Nathan Wilson B.Sc., Rockhurst University, 2010 Submitted to the graduate degree program in Anatomy and Cell Biology and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Dr. Irfan Saadi, Ph.D. (Chair) Dr. András Czirók, Ph.D. Dr. William Kinsey, Ph.D. Dr. Paul Trainor, Ph.D. Dr. Pamela Tran, Ph.D. Dr. Jinxi Wang, M.D., Ph.D. Date Defended: 19 April 2017 The dissertation committee for Nathan Wilson certifies that this is the approved version of the following dissertation: Characterization of SPECC1L function in cell adhesion and migration in craniofacial morphogenesis Dr. Irfan Saadi, Ph.D. (Chair) Date approved: 12 May 2017 ii Abstract Orofacial clefts are frequent congenital malformations, which can result from reduced contribution of cranial neural crest cells (CNCCs) to the developing cranium. Upon delamination from embryonic neural folds, CNCCs migrate to pharyngeal arches, which give rise to mid-facial structures. Previously, mutations in the cytoskeletal gene SPECC1L were implicated in rare, severe atypical facial clefting. Overexpression of a patient isolated SPECC1L mutation showed disrupted association with acetylated microtubules. We show a similar disruption upon overexpression of SPECC1L variants isolated from patients with less severe syndromic cleft lip and palate. Severe Specc1l deficiency in homozygous mouse mutants is embryonic lethal, showing a reduction in pan-AKT levels and arrested CNCC delamination from the neural folds. Staining of adherens junction (AJ) proteins is increased in mutant neural folds, consistent with impaired CNCC delamination, a process requiring AJ dissolution. -

Mitotic Checkpoints and Chromosome Instability Are Strong Predictors of Clinical Outcome in Gastrointestinal Stromal Tumors
MITOTIC CHECKPOINTS AND CHROMOSOME INSTABILITY ARE STRONG PREDICTORS OF CLINICAL OUTCOME IN GASTROINTESTINAL STROMAL TUMORS. Pauline Lagarde1,2, Gaëlle Pérot1, Audrey Kauffmann3, Céline Brulard1, Valérie Dapremont2, Isabelle Hostein2, Agnès Neuville1,2, Agnieszka Wozniak4, Raf Sciot5, Patrick Schöffski4, Alain Aurias1,6, Jean-Michel Coindre1,2,7 Maria Debiec-Rychter8, Frédéric Chibon1,2. Supplemental data NM cases deletion frequency. frequency. deletion NM cases Mand between difference the highest setswith of theprobe a view isdetailed panel Bottom frequently. sorted totheless deleted theprobe are frequently from more and thefrequency deletion represent Yaxes inblue. are cases (NM) metastatic for non- frequencies Corresponding inmetastatic (red). probe (M)cases sets figureSupplementary 1: 100 100 20 40 60 80 20 40 60 80 0 0 chr14 1 chr14 88 chr14 175 chr14 262 chr9 -MTAP 349 chr9 -MTAP 436 523 chr9-CDKN2A 610 Histogram presenting the 2000 more frequently deleted deleted frequently the 2000 more presenting Histogram chr9-CDKN2A 697 chr9-CDKN2A 784 chr9-CDKN2B 871 chr9-CDKN2B 958 chr9-CDKN2B 1045 chr22 1132 chr22 1219 chr22 1306 chr22 1393 1480 1567 M NM 1654 1741 1828 1915 M NM GIST14 GIST2 GIST16 GIST3 GIST19 GIST63 GIST9 GIST38 GIST61 GIST39 GIST56 GIST37 GIST47 GIST58 GIST28 GIST5 GIST17 GIST57 GIST47 GIST58 GIST28 GIST5 GIST17 GIST57 CDKN2A Supplementary figure 2: Chromosome 9 genomic profiles of the 18 metastatic GISTs (upper panel). Deletions and gains are indicated in green and red, respectively; and color intensity is proportional to copy number changes. A detailed view is given (bottom panel) for the 6 cases presenting a homozygous 9p21 deletion targeting CDKN2A locus (dark green). -

A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression
Supplementary Materials A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression Table S1. Statistically significant DEGs (Adj. p-value < 0.01) derived from meta-analysis for samples irradiated with high doses of HZE particles, collected 6-24 h post-IR not common with any other meta- analysis group. This meta-analysis group consists of 3 DEG lists obtained from DGEA, using a total of 11 control and 11 irradiated samples [Data Series: E-MTAB-5761 and E-MTAB-5754]. Ensembl ID Gene Symbol Gene Description Up-Regulated Genes ↑ (2425) ENSG00000000938 FGR FGR proto-oncogene, Src family tyrosine kinase ENSG00000001036 FUCA2 alpha-L-fucosidase 2 ENSG00000001084 GCLC glutamate-cysteine ligase catalytic subunit ENSG00000001631 KRIT1 KRIT1 ankyrin repeat containing ENSG00000002079 MYH16 myosin heavy chain 16 pseudogene ENSG00000002587 HS3ST1 heparan sulfate-glucosamine 3-sulfotransferase 1 ENSG00000003056 M6PR mannose-6-phosphate receptor, cation dependent ENSG00000004059 ARF5 ADP ribosylation factor 5 ENSG00000004777 ARHGAP33 Rho GTPase activating protein 33 ENSG00000004799 PDK4 pyruvate dehydrogenase kinase 4 ENSG00000004848 ARX aristaless related homeobox ENSG00000005022 SLC25A5 solute carrier family 25 member 5 ENSG00000005108 THSD7A thrombospondin type 1 domain containing 7A ENSG00000005194 CIAPIN1 cytokine induced apoptosis inhibitor 1 ENSG00000005381 MPO myeloperoxidase ENSG00000005486 RHBDD2 rhomboid domain containing 2 ENSG00000005884 ITGA3 integrin subunit alpha 3 ENSG00000006016 CRLF1 cytokine receptor like -
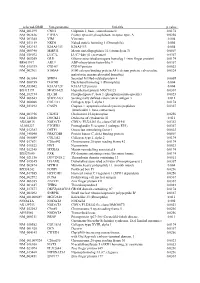
Supplementary Figures S1-S3
selected-GBID Uni-genename Uni-title p value NM_001299 CNN1 Calponin 1, basic, smooth muscle 0.0174 NM_002836 PTPRA Protein tyrosine phosphatase, receptor type, A 0.0256 NM_003380 VIM Vimentin 0.004 NM_033119 NKD1 Naked cuticle homolog 1 (Drosophila) 0.004 NM_052913 KIAA1913 KIAA1913 0.004 NM_005940 MMP11 Matrix metallopeptidase 11 (stromelysin 3) 0.0069 NM_018032 LUC7L LUC7-like (S. cerevisiae) 0.0367 NM_005269 GLI1 Glioma-associated oncogene homolog 1 (zinc finger protein) 0.0174 BE463997 ARL9 ADP-ribosylation factor-like 9 0.0367 NM_015939 CGI-09 CGI-09 protein 0.0023 NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, 0.0324 metastasin, murine placental homolog) NM_003014 SFRP4 Secreted frizzled-related protein 4 0.0005 NM_080759 DACH1 Dachshund homolog 1 (Drosophila) 0.004 NM_053042 KIAA1729 KIAA1729 protein 0.004 BX415194 MGC16121 Hypothetical protein MGC16121 0.0367 NM_182734 PLCB1 Phospholipase C, beta 1 (phosphoinositide-specific) 0.0023 NM_006643 SDCCAG3 Serologically defined colon cancer antigen 3 0.011 NM_000088 COL1A1 Collagen, type I, alpha 1 0.0174 NM_033292 CASP1 Caspase 1, apoptosis-related cysteine peptidase 0.0367 (interleukin 1, beta, convertase) NM_003956 CH25H Cholesterol 25-hydroxylase 0.0256 NM_144658 DOCK11 Dedicator of cytokinesis 11 0.011 AK024935 NODATA CDNA: FLJ21283 fis, clone COL01910 0.0363 AL050227 PTGER3 Prostaglandin E receptor 3 (subtype EP3) 0.0367 NM_012383 OSTF1 Osteoclast stimulating factor 1 0.0023 NM_145040 PRKCDBP Protein kinase C, delta binding protein 0.0069 NM_000089 -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

Novel and Highly Recurrent Chromosomal Alterations in Se´Zary Syndrome
Research Article Novel and Highly Recurrent Chromosomal Alterations in Se´zary Syndrome Maarten H. Vermeer,1 Remco van Doorn,1 Remco Dijkman,1 Xin Mao,3 Sean Whittaker,3 Pieter C. van Voorst Vader,4 Marie-Jeanne P. Gerritsen,5 Marie-Louise Geerts,6 Sylke Gellrich,7 Ola So¨derberg,8 Karl-Johan Leuchowius,8 Ulf Landegren,8 Jacoba J. Out-Luiting,1 Jeroen Knijnenburg,2 Marije IJszenga,2 Karoly Szuhai,2 Rein Willemze,1 and Cornelis P. Tensen1 Departments of 1Dermatology and 2Molecular Cell Biology, Leiden University Medical Center, Leiden, the Netherlands; 3Department of Dermatology, St Thomas’ Hospital, King’s College, London, United Kingdom; 4Department of Dermatology, University Medical Center Groningen, Groningen, the Netherlands; 5Department of Dermatology, Radboud University Nijmegen Medical Center, Nijmegen, the Netherlands; 6Department of Dermatology, Gent University Hospital, Gent, Belgium; 7Department of Dermatology, Charite, Berlin, Germany; and 8Department of Genetics and Pathology, Rudbeck Laboratory, University of Uppsala, Uppsala, Sweden Abstract Introduction This study was designed to identify highly recurrent genetic Se´zary syndrome (Sz) is an aggressive type of cutaneous T-cell alterations typical of Se´zary syndrome (Sz), an aggressive lymphoma/leukemia of skin-homing, CD4+ memory T cells and is cutaneous T-cell lymphoma/leukemia, possibly revealing characterized by erythroderma, generalized lymphadenopathy, and pathogenetic mechanisms and novel therapeutic targets. the presence of neoplastic T cells (Se´zary cells) in the skin, lymph High-resolution array-based comparative genomic hybridiza- nodes, and peripheral blood (1). Sz has a poor prognosis, with a tion was done on malignant T cells from 20 patients. disease-specific 5-year survival of f24% (1). -

Whole-Exome Sequencing Identified Genetic Risk Factors for Asparaginase-Related Complications in Childhood ALL Patients
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 27), pp: 43752-43767 Research Paper Whole-exome sequencing identified genetic risk factors for asparaginase-related complications in childhood ALL patients Rachid Abaji1,7, Vincent Gagné1, Chang Jiang Xu1, Jean-François Spinella1, Francesco Ceppi1, Caroline Laverdière1,2, Jean-Marie Leclerc1,2, Stephen E. Sallan3,4, Donna Neuberg5, Jeffery L. Kutok6, Lewis B. Silverman3,4, Daniel Sinnett1,2 and Maja Krajinovic1,2,7 1 Research Center, Centre Hospitalier Universitaire Sainte-Justine, Montreal, QC, Canada 2 Department of Pediatrics, University of Montreal, Montreal, QC, Canada 3 Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA, USA 4 Division of Hematology/Oncology, Children’s Hospital, Boston, MA, USA 5 Department of Biostatistics and Computational Biology, Dana-Farber Cancer Institute, Boston, MA, USA 6 Department of Pathology, Brigham and Women’s Hospital, Boston, MA, USA 7 Department of Pharmacology, University of Montreal, Montreal, QC, Canada Correspondence to: Maja Krajinovic, email: [email protected] Keywords: acute lymphoblastic leukemia; asparaginase; exome-wide association; pharmacogenetics; whole-exome sequencing Received: April 21, 2017 Accepted: April 27, 2017 Published: May 17, 2017 Copyright: Abaji et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Allergy, pancreatitis and thrombosis are common side-effects of childhood acute lymphoblastic leukemia (ALL) treatment that are associated with the use of asparaginase (ASNase), a key component in most ALL treatment protocols. -

Comparative Genomic Analysis of Rapidly Evolving SARS-Cov-2 Viruses 2 Reveal Mosaic Pattern of Phylogeographical Distribution
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.25.006213; this version posted April 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Comparative Genomic Analysis of Rapidly Evolving SARS-CoV-2 Viruses 2 Reveal Mosaic Pattern of Phylogeographical Distribution 3 Roshan Kumar1, Helianthous Verma2, Nirjara Singhvi3, Utkarsh Sood4, Vipin Gupta5, Mona 4 Singh5, Rashmi Kumari6, Princy Hira7, Shekhar Nagar3, Chandni Talwar3, Namita Nayyar8, 5 Shailly Anand9, Charu Dogra Rawat2, Mansi Verma8, Ram Krishan Negi3, Yogendra Singh3 6 and Rup Lal4* 7 8 9 10 Authors Affiliations 11 1P.G. Department of Zoology, Magadh University, Bodh Gaya, Bihar-824234, India 12 2Department of Zoology, Ramjas College, University of Delhi, New Delhi-110007, India 13 3Department of Zoology, University of Delhi, New Delhi-110007, India 14 4The Energy and Resources Institute, Darbari Seth Block, IHC Complex, Lodhi Road, New 15 Delhi-110003, India 16 5PhiXGen Private Limited, Gurugram, Haryana 122001, India 17 6Department of Zoology, College of Commerce, Arts & Science, Patliputra University, Patna, 18 Bihar-800020, India 19 7Department of Zoology, Maitreyi College, University of Delhi, New Delhi-110021, India 20 8Department of Zoology, Sri Venkateswara College, University of Delhi, New Delhi-110021, 21 India 22 9Department of Zoology, Deen Dayal Upadhyaya College, University of Delhi, New Delhi- 23 110078, India 24 25 26 27 28 *Corresponding Author 29 Email: [email protected] 30 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.03.25.006213; this version posted April 16, 2020. -

WO 2015/149034 A2 1 October 2015 (01.10.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/149034 A2 1 October 2015 (01.10.2015) P O P C T (51) International Patent Classification: (71) Applicant: LIFE TECHNOLOGIES CORPORATION C12Q 1/68 (2006.01) [US/US]; c/o IP Legal Department Docketing, 5791 Van Allen Way, Carlsbad, California 92008 (US). (21) International Application Number: PCT/US2015/023 197 (72) Inventors: RHODES, Daniel; Life Technologies Corpora tion, c/o IP Legal Department Docketing, 579 1 Van Allen (22) International Filing Date: Way, Carlsbad, California 92008 (US). SADIS, Seth; Life 27 March 2015 (27.03.2015) Technologies Corporation, c/o IP Legal Department Dock (25) Filing Language: English eting, 5791 Van Allen Way, Carlsbad, California 92008 (US). WYNGAARD, Peter; Life Technologies Corpora (26) Publication Language: English tion, c/o IP Legal Department Docketing, 579 1 Van Allen (30) Priority Data: Way, Carlsbad, California 92008 (US). KHAZANOV, 61/971,455 27 March 2014 (27.03.2014) US Nikolay; Life Technologies Corporation, c/o IP Legal De 61/993,732 15 May 2014 (15.05.2014) US partment Docketing, 5791 Van Allen Way, Carlsbad, Cali 62/004,727 29 May 2014 (29.05.2014) us fornia 92008 (US). BANDLA, Santhoshi; Life Technolo 62/092,898 17 December 2014 (17. 12.2014) us gies Corporation, c/o IP Legal Department Docketing, 5791 Van Allen Way, Carlsbad, California 92008 (US). [Continued on nextpage] (54) Title: GENE FUSIONS AND GENE VARIANTS ASSOCIATED WITH CANCER (57) Abstract: The disclosure provides gene fusions, gene variants, and novel as RNASeq sociations with disease states, as well as kits, probes, and methods of using the (.bam) same. -

Chromosomal Microarray Analysis in the Genetic Evaluation of 279
D’Angelo et al. Molecular Cytogenetics (2018) 11:14 DOI 10.1186/s13039-018-0363-7 RESEARCH Open Access Chromosomal microarray analysis in the genetic evaluation of 279 patients with syndromic obesity Carla Sustek D’Angelo1*, Monica Castro Varela1, Claudia Irene Emílio de Castro1, Paulo Alberto Otto1, Ana Beatriz Alvarez Perez2, Charles Marques Lourenço3, Chong Ae Kim4, Debora Romeo Bertola4, Fernando Kok5, Luis Garcia-Alonso2 and Celia Priszkulnik Koiffmann1 Abstract Background: Syndromic obesity is an umbrella term used to describe cases where obesity occurs with additional phenotypes. It often arises as part of a distinct genetic syndrome with Prader-Willi syndrome being a classical example. These rare forms of obesity provide a unique source for identifying obesity-related genetic changes. Chromosomal microarray analysis (CMA) has allowed the characterization of new genetic forms of syndromic obesity, which are due to copy number variants (CNVs); however, CMA in large cohorts requires more study. The aim of this study was to characterize the CNVs detected by CMA in 279 patients with a syndromic obesity phenotype. Results: Pathogenic CNVs were detected in 61 patients (22%) and, among them, 35 had overlapping/recurrent CNVs. Genomic imbalance disorders known to cause syndromic obesity were found in 8.2% of cases, most commonly deletions of 1p36, 2q37 and 17p11.2 (5.4%), and we also detected deletions at 1p21.3, 2p25.3, 6q16, 9q34, 16p11.2 distal and proximal, as well as an unbalanced translocation resulting in duplication of the GNB3 gene responsible for a syndromic for of childhood obesity. Deletions of 9p terminal and 22q11.2 proximal/distal were found in 1% and 3% of cases, respectively.