Intrahepatic Biliary Dilatation with Aortoarteritis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Journal of Medical Genetics April 1992 Vol 29 No4 Contents Original Articles
Journal of Medical Genetics April 1992 Vol 29 No4 Contents Original articles Beckwith-Wiedemann syndrome: a demonstration of the mechanisms responsible for the excess J Med Genet: first published as on 1 April 1992. Downloaded from of transmitting females C Moutou, C Junien, / Henry, C Bonai-Pellig 217 Evidence for paternal imprinting in familial Beckwith-Wiedemann syndrome D Viljoen, R Ramesar 221 Sex reversal in a child with a 46,X,Yp+ karyotype: support for the existence of a gene(s), located in distal Xp, involved in testis formation T Ogata, J R Hawkins, A Taylor, N Matsuo, J-1 Hata, P N Goodfellow 226 Highly polymorphic Xbol RFLPs of the human 21 -hydroxylase genes among Chinese L Chen, X Pan, Y Shen, Z Chen, Y Zhang, R Chen 231 Screening of microdeletions of chromosome 20 in patients with Alagille syndrome C Desmaze, J F Deleuze, A M Dutrillaux, G Thomas, M Hadchouel, A Aurias 233 Confirmation of genetic linkage between atopic IgE responses and chromosome 1 1 ql 3 R P Young, P A Sharp, J R Lynch, J A Faux, G M Lathrop, W 0 C M Cookson, J M Hopkini 236 Age at onset and life table risks in genetic counselling for Huntington's disease P S Harper, R G Newcombe 239 Genetic and clinical studies in autosomal dominant polycystic kidney disease type 1 (ADPKD1) E Coto, S Aguado, J Alvarez, M J Menendez-DIas, C Lopez-Larrea 243 Short communication Evidence for linkage disequilibrium between D16S94 and the adult onset polycystic kidney disease (PKD1) gene S E Pound, A D Carothers, P M Pignatelli, A M Macnicol, M L Watson, A F Wright 247 Technical note A strategy for the rapid isolation of new PCR based DNA polymorphisms P R Hoban, M F Santibanez-Koref, J Heighway 249 http://jmg.bmj.com/ Case reports Campomelic dysplasia associated with a de novo 2q;1 7q reciprocal translocation I D Young, J M Zuccollo, E L Maltby, N J Broderick 251 A complex chromosome rearrangement with 10 breakpoints: tentative assignment of the locus for Williams syndrome to 4q33-q35.1 R Tupler, P Maraschio, A Gerardo, R Mainieri G Lanzi L Tiepolo 253 on September 26, 2021 by guest. -

Chromosome 20
Chromosome 20 ©Chromosome Disorder Outreach Inc. (CDO) Technical genetic content provided by Dr. Iosif Lurie, M.D. Ph.D Medical Geneticist and CDO Medical Consultant/Advisor. Ideogram courtesy of the University of Washington Department of Pathology: ©1994 David Adler.hum_20.gif Introduction Chromosome 20 contains about 2% of the whole genetic material. Its genetic length is ~63 Mb. The long arm (~36 Mb) is a little bit larger than the short arm (~27 Mb). Chromosome 20 contains ~700–800 genes. Less than 10% of these genes are known to be related to human diseases. Deletions or duplications of these genes, which may be found in patients with chromosomal abnormalities, cause mostly functional defects, including a delay of psycho–motor development and seizures. Only a few genes may lead (when deleted) to structural defects of the heart, liver, extremities and other organs. Deletions of Chromosome 20 There is a relatively small number of known conditions caused by deletions and duplications of various segments of chromosome 20. Almost all of these deletions and duplications became recognized after usage of molecular cytogenetics. Only a handful of reports on patients with these abnormalities were available only 10 years ago. Because these methods open wide an opportunity to examine abnormalities of this previously not–well studied chromosome, there are no doubts that some new syndromes caused by deletions (or duplications) of chromosome 20 will be delineated in the near future. Currently, the most frequent forms of chromosome 20 deletions are deletions 20p12, involving the JAG1 gene and Alagille syndrome, and deletions 20q13.13q13.2, involving the SALL4 gene. -

Its Place Among Other Genetic Causes of Renal Disease
J Am Soc Nephrol 13: S126–S129, 2002 Anderson-Fabry Disease: Its Place among Other Genetic Causes of Renal Disease JEAN-PIERRE GRU¨ NFELD,* DOMINIQUE CHAUVEAU,* and MICHELINE LE´ VY† *Service of Nephrology, Hoˆpital Necker, Paris, France; †INSERM U 535, Baˆtiment Gregory Pincus, Kremlin- Biceˆtre, France. In the last two decades, decisive advances have been made in Nephropathic cystinosis, first described in 1903, is an auto- the field of human genetics, including renal genetics. The somal recessive disorder characterized by the intra-lysosomal responsible genes have been mapped and then identified in accumulation of cystine. It is caused by a defect in the transport most monogenic renal disorders by using positional cloning of cystine out of the lysosome, a process mediated by a carrier and/or candidate gene approaches. These approaches have that remained unidentified for several decades. However, an been extremely efficient since the number of identified genetic important management step was devised in 1976, before the diseases has increased exponentially over the last 5 years. The biochemical defect was characterized in 1982. Indeed cysteam- data derived from the Human Genome Project will enable a ine, an aminothiol, reacts with cystine to form cysteine-cys- more rapid identification of the genes involved in the remain- teamine mixed disulfide that can readily exit the cystinotic ing “orphan” inherited renal diseases, provided their pheno- lysosome. This drug, if used early and in high doses, retards the types are well characterized. We have entered the post-gene progression of cystinosis in affected subjects by reducing intra- era. What is/are the function(s) of these genes? What are the lysosomal cystine concentrations. -

Guideline for the Evaluation of Cholestatic Jaundice
CLINICAL GUIDELINES Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition ÃRima Fawaz, yUlrich Baumann, zUdeme Ekong, §Bjo¨rn Fischler, jjNedim Hadzic, ôCara L. Mack, #Vale´rie A. McLin, ÃÃJean P. Molleston, yyEzequiel Neimark, zzVicky L. Ng, and §§Saul J. Karpen ABSTRACT Cholestatic jaundice in infancy affects approximately 1 in every 2500 term PREAMBLE infants and is infrequently recognized by primary providers in the setting of holestatic jaundice in infancy is an uncommon but poten- physiologic jaundice. Cholestatic jaundice is always pathologic and indicates tially serious problem that indicates hepatobiliary dysfunc- hepatobiliary dysfunction. Early detection by the primary care physician and tion.C Early detection of cholestatic jaundice by the primary care timely referrals to the pediatric gastroenterologist/hepatologist are important physician and timely, accurate diagnosis by the pediatric gastro- contributors to optimal treatment and prognosis. The most common causes of enterologist are important for successful treatment and an optimal cholestatic jaundice in the first months of life are biliary atresia (25%–40%) prognosis. The Cholestasis Guideline Committee consisted of 11 followed by an expanding list of monogenic disorders (25%), along with many members of 2 professional societies: the North American Society unknown or multifactorial (eg, parenteral nutrition-related) causes, each of for Gastroenterology, Hepatology and Nutrition, and the European which may have time-sensitive and distinct treatment plans. Thus, these Society for Gastroenterology, Hepatology and Nutrition. This guidelines can have an essential role for the evaluation of neonatal cholestasis committee has responded to a need in pediatrics and developed to optimize care. -

Focal Liver Hyperplasia in Alagille Syndrome: Assessment with Hepatoreceptor and Hepatobiliary Imaging
Focal Liver Hyperplasia in Alagille Syndrome: Assessment with Hepatoreceptor and Hepatobiliary Imaging Tatsuo Torizuka, Nagara Tamaki, Toru Fujita, Yoshiharu Yonekura, Shinji Uemoto, Koichi Tanaka, Yoshio Yamaoka and Junj i Konishi Department of Nuclear Medicine and Second Department of Surgery, Kyoto University Faculty of Medicine, Kyoto, Japan manifestations of Alagille syndrome, including hypertelorism, A child with Alagille syndrome, characterized by intrahepatic bile broad forehead, high nose and pointed chin. The patient's mother duct paucity, developed severe liver cirrhosis and was referred for liver transplantation. In the pre-transplantation evaluation, scinti- did not have the same facial features. graphic scans were performed using 99mTc-galactosyl serum albu Cholecystectomy was performed for cholestasis when the patient min (""Tc-GSA) as a hepatoreceptor binding agent and 99mTc- was 5 yr old. At 6 yr, severe liver cirrhosis was suspected based on pyridoxyl-5-methyl-tryptophan (""Tc-PMT) as a hepatobiliary serum biochemical data. The serum direct bilirubin value was 21.5 agent. These studies demonstrated severe hepatobiliary dysfunc mg/dl. Jaundice and marked venous dilatation on the abdominal tion with an area of increased focal uptake in the liver. Histological wall were observed. X-ray CT images revealed liver atrophy with examination at surgery confirmed that this focal lesion was an area splenomegaly and a high density nodular lesion in the medial right of compensatory hyperplasia in advanced biliary cirrhosis. We lobe of the liver (Fig. 1). present the usefulness of these tracers for detecting the focal The patient underwent dynamic 99Tc-GSA imaging under a hyperplasia of the liver. rotating gamma camera. -

Essential Genetics 5
Essential genetics 5 Disease map on chromosomes 例 Gaucher disease 単一遺伝子病 天使病院 Prader-Willi syndrome 隣接遺伝子症候群,欠失が主因となる疾患 臨床遺伝診療室 外木秀文 Trisomy 13 複数の遺伝子の重複によって起こる疾患 挿画 Koromo 遺伝子の座位あるいは欠失等の範囲を示す Copyright (c) 2010 Social Medical Corporation BOKOI All Rights Reserved. Disease map on chromosome 1 Gaucher disease Chromosome 1q21.1 1p36 deletion syndrome deletion syndrome Adrenoleukodystrophy, neonatal Cardiomyopathy, dilated, 1A Zellweger syndrome Charcot-Marie-Tooth disease Emery-Dreifuss muscular Hypercholesterolemia, familial dystrophy Hutchinson-Gilford progeria Ehlers-Danlos syndrome, type VI Muscular dystrophy, limb-girdle type Congenital disorder of Insensitivity to pain, congenital, glycosylation, type Ic with anhidrosis Diamond-Blackfan anemia 6 Charcot-Marie-Tooth disease Dejerine-Sottas syndrome Marshall syndrome Stickler syndrome, type II Chronic granulomatous disease due to deficiency of NCF-2 Alagille syndrome 2 Copyright (c) 2010 Social Medical Corporation BOKOI All Rights Reserved. Disease map on chromosome 2 Epiphyseal dysplasia, multiple Spondyloepimetaphyseal dysplasia Brachydactyly, type D-E, Noonan syndrome Brachydactyly-syndactyly syndrome Peters anomaly Synpolydactyly, type II and V Parkinson disease, familial Leigh syndrome Seizures, benign familial Multiple pterygium syndrome neonatal-infantile Escobar syndrome Ehlers-Danlos syndrome, Brachydactyly, type A1 type I, III, IV Waardenburg syndrome Rhizomelic chondrodysplasia punctata, type 3 Alport syndrome, autosomal recessive Split-hand/foot malformation Crigler-Najjar -

Outcome of Liver Disease in Children with Alagille Syndrome: a Study of 163 Patients Gut: First Published As 10.1136/Gut.49.3.431 on 1 September 2001
Gut 2001;49:431–435 431 Outcome of liver disease in children with Alagille syndrome: a study of 163 patients Gut: first published as 10.1136/gut.49.3.431 on 1 September 2001. Downloaded from P Lykavieris, M Hadchouel, C Chardot, O Bernard Abstract to have a relatively good long term prognosis in Background and aims—Various opinions terms of liver disease45; however, it is now well have been expressed as to the long term recognised that some patients with AGS can prognosis of liver disease associated with present with severe complications of liver Alagille syndrome (AGS). disease.6–12 We therefore reviewed the charts of Patients and methods—We reviewed the 174 patients with AGS presenting in childhood outcome of 163 children with AGS and to evaluate the role of the liver condition in liver involvement, investigated from 1960 mortality, morbidity, and long term outcome. to 2000, the end point of the study (median age 10 years (range 2 months to 44 years)) being death, liver transplantation, or the Patients and methods last visit. One hundred and seventy four children with Results—At the study end point, of the 132 AGS (106 boys) were investigated at Bicêtre patients who presented with neonatal Hospital between 1960 and 2000. Twenty four cholestatic jaundice, 102 remained jaun- had a sibling aVected by AGS; seven of these diced, 112 had poorly controlled pruritus, siblings are included in this series as well as two and 40 had xanthomas; cirrhosis was oVspring of aVected mothers. All patients had found in 35/76 livers, varices in 25/71 at least three of the five major clinical features. -

EUROCAT Syndrome Guide
JRC - Central Registry european surveillance of congenital anomalies EUROCAT Syndrome Guide Definition and Coding of Syndromes Version July 2017 Revised in 2016 by Ingeborg Barisic, approved by the Coding & Classification Committee in 2017: Ester Garne, Diana Wellesley, David Tucker, Jorieke Bergman and Ingeborg Barisic Revised 2008 by Ingeborg Barisic, Helen Dolk and Ester Garne and discussed and approved by the Coding & Classification Committee 2008: Elisa Calzolari, Diana Wellesley, David Tucker, Ingeborg Barisic, Ester Garne The list of syndromes contained in the previous EUROCAT “Guide to the Coding of Eponyms and Syndromes” (Josephine Weatherall, 1979) was revised by Ingeborg Barisic, Helen Dolk, Ester Garne, Claude Stoll and Diana Wellesley at a meeting in London in November 2003. Approved by the members EUROCAT Coding & Classification Committee 2004: Ingeborg Barisic, Elisa Calzolari, Ester Garne, Annukka Ritvanen, Claude Stoll, Diana Wellesley 1 TABLE OF CONTENTS Introduction and Definitions 6 Coding Notes and Explanation of Guide 10 List of conditions to be coded in the syndrome field 13 List of conditions which should not be coded as syndromes 14 Syndromes – monogenic or unknown etiology Aarskog syndrome 18 Acrocephalopolysyndactyly (all types) 19 Alagille syndrome 20 Alport syndrome 21 Angelman syndrome 22 Aniridia-Wilms tumor syndrome, WAGR 23 Apert syndrome 24 Bardet-Biedl syndrome 25 Beckwith-Wiedemann syndrome (EMG syndrome) 26 Blepharophimosis-ptosis syndrome 28 Branchiootorenal syndrome (Melnick-Fraser syndrome) 29 CHARGE -

Congenital Hypothyroidism: Update and Perspectives
6 179 C Peters and others Congenital hypothyroidism: 179:6 R297–R317 Review update DIAGNOSIS OF ENDOCRINE DISEASE Congenital hypothyroidism: update and perspectives C Peters1, A S P van Trotsenburg2 and N Schoenmakers3 1Department of Endocrinology, Great Ormond Street Hospital for Children, London, UK, 2Emma Children’s, Correspondence Amsterdam UMC, University of Amsterdam, Pediatric Endcorinology, Amsterdam, the Netherlands, and 3University should be addressed of Cambridge Metabolic Research Laboratories, Wellcome Trust-Medical Research Council Institute of Metabolic to N Schoenmakers Science, Addenbrooke’s Hospital, Cambridge, UK Email [email protected] Abstract Congenital hypothyroidism (CH) may be primary, due to a defect affecting the thyroid gland itself, or central, due to impaired thyroid-stimulating hormone (TSH)-mediated stimulation of the thyroid gland as a result of hypothalamic or pituitary pathology. Primary CH is the most common neonatal endocrine disorder, traditionally subdivided into thyroid dysgenesis (TD), referring to a spectrum of thyroid developmental abnormalities, and dyshormonogenesis, where a defective molecular pathway for thyroid hormonogenesis results in failure of hormone production by a structurally intact gland. Delayed treatment of neonatal hypothyroidism may result in profound neurodevelopmental delay; therefore, CH is screened for in developed countries to facilitate prompt diagnosis. Central congenital hypothyroidism (CCH) is a rarer entity which may occur in isolation, or (more frequently) in association with additional pituitary hormone deficits. CCH is most commonly defined biochemically by failure of appropriate TSH elevation despite subnormal thyroid hormone levels and will therefore evade diagnosis in primary, TSH-based CH-screening programmes. This review will discuss recent genetic aetiological advances in CH and summarize epidemiological data and clinical diagnostic challenges, focussing on primary CH and isolated CCH. -

Alagille Syndrome a Guide
Alagille Syndrome A Guide An explanation of what Alagille syndrome is, its causes, diagnosis and treatment Alagille Syndrome Alagille Syndrome What is Alagille syndrome? ....................................... 3 This information has primarily been written for: § Parents/carers of children with Alagille syndrome How many children are affected by Alagille syndrome? ................................................................. 4 Others may also find this information useful: What are the features of Alagille syndrome? ............. 4 § Young people with Alagille syndrome § Healthcare professionals who would like to find out more What causes Alagille syndrome? .............................. 6 about the condition How is Alagille syndrome diagnosed? ...................... 8 It provides information on: What are the effects of Alagille syndrome? ............... 9 § What Alagille syndrome is How is Alagille syndrome treated? .......................... 11 § Causes § Diagnosis § Treatment You may also find it helpful to read the following CLDF leaflets: § An Introduction to Liver Disease § Pruritus (itch) § Portal hypertension and ascites What is Alagille syndrome? Alagille syndrome is a rare, genetic condition. It can affect different parts of the body including the liver, heart, kidneys, eyes, face and bones. 2 3 Alagille Syndrome Alagille Syndrome How many children are affected by Diagram of the liver and surrounding organs Alagille syndrome? Liver Alagille syndrome affects around one in every 30,000 live births. Bile ducts – those with Alagille syndrome may What are the features of Alagille have fewer than normal syndrome? Gallbladder Pancreas There are many different ways Alagille syndrome can affect an individual. It differs from person to person and even two people Duodenum in the same family with Alagille syndrome can have different Jejunum features and symptoms. Some people have a very mild form of the condition and reach adulthood without knowing they have Alagille syndrome. -
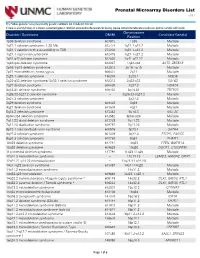
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome -

Genetic Basis of Human Congenital Heart Disease
This is a free sample of content from Heart Development and Disease. Click here for more information on how to buy the book. Genetic Basis of Human Congenital Heart Disease Shannon N. Nees1 and Wendy K. Chung1,2 1Department of Pediatrics,2Department of Medicine, Columbia University Irving Medical Center, New York, New York 10032, USA Correspondence: [email protected] Congenital heart disease (CHD) is the most common major congenital anomaly with an incidence of ∼1% of live births and is a significant cause of birth defect–related mortality. The genetic mechanisms underlying the development of CHD are complex and remain incompletely understood. Known genetic causes include all classes of genetic variation in- cluding chromosomal aneuploidies, copy number variants, and rare and common single- nucleotide variants, which can be either de novo or inherited. Among patients with CHD, ∼8%–12% have a chromosomal abnormality or aneuploidy, between 3% and 25% have a copy number variation, and 3%–5% have a single-gene defect in an established CHD gene with higher likelihood of identifying a genetic cause in patients with nonisolated CHD. These genetic variants disrupt or alter genes that play an important role in normal cardiac develop- ment and in some cases have pleiotropic effects on other organs. This work reviews some of the most common genetic causes of CHD as well as what is currently known about the underlying mechanisms. ongenital heart disease (CHD) is the most are underdeveloped left-sided cardiac structures Ccommon major congenital anomaly with an and only a single functioning ventricle. The high incidence of ∼1% of live births (Hoffman and concordance in monozygotic twins, the in- Kaplan 2002; Calzolari et al.