Bioenergetic Impairment in Congenital Muscular Dystrophy Type 1A And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Synthesis and Evaluation of Chromone Derivatives As Inhibitors of Monoamine Oxidase
Synthesis and evaluation of chromone derivatives as inhibitors of monoamine oxidase AN Mpitimpiti 21253005 Dissertation submitted in fulfilment of the requirements for the degree Magister Scientiae in Pharmaceutical Chemistry at the Potchefstroom Campus of the North-West University Supervisor: Dr ACU Lourens Co-supervisors: Dr A Petzer Prof JP Petzer November 2014 The financial assistance of the National Research Foundation (NRF) and the Medical Research Council (MRC) towards this research is hereby acknowledged. Opinions expressed and conclusions arrived at are those of the author and are not necessarily to be attributed to the NRF or MRC. Acknowledgements • All glory be to God. • I am deeply indebted to the following for their immense support and contribution: o My supervisor - Dr A.C.U Lourens (your guidance is truly boundless). o My co-supervisors, Prof J.P Petzer and Dr A. Petzer. o Dr J.Jordaan. • My mother, Magret Chigeza (for your unconditional love and support). • My family and friends. • All those who knowingly and unknowingly supported me through out this journey. Psalm 115 verse 1: ‘Not to us, O Lord, not to us, but to Your Name be the glory, because of Your love and faithfulness’ i Table of Contents Abstract ...........................................................................................................................iv Opsomming ....................................................................................................................vii List of abbreviations .......................................................................................................xi -

Recent Research Developments in Parkinson's Disease
Page 12 to 30 Current Opinions in Neurological Science ISSN: 2575-5447 Review Article Volume 5 Issue 1 • 2020 Recent Research Developments in Parkinson’s Disease Alain L. Fymat Institute Professor, International Institute of Medicine & Science, California, U.S.A Received : February 09, 2020 Published : February 24, 2020 *Corresponding Author: Alain L. Fymat, Professor, International Institute of Medicine & Science, California, U.S.A Copyright © All rights are reserved by Alain L. Fymat. Abstract Important research developments have recently occurred in the understanding of Parkinson’s mechanisms and the symptomatic therapy of the disease. This article reviews such developments and their place in the disease’s therapeutic armamentarium. It also assesses their potential contribution(s) to our search for the root cause(s) of the disease. Improved symptomatic therapy has taken place for both motor and non-motor systems with the progressive development of several therapeutic drugs. This article also explores new research vistas including the role of gene therapy in the search for Parkinson’s disease-modifying therapy and neuroprotective treatments such as neural transplantation, stem cell transplantation, and transcranial magnetic stimulation. In this context, our two interacting brains (brain-in-the-skull, brain-in-the-gut) should underpin Braak’s hypothesis as to the origin of Parkinson’s. In the search for disease-modifying therapies, the article reviews the two therapeutic strategies currently followed, the correlation between a reduced occurrence or prevalence (or both) of PD and the consumption of compounds such as caffeine or nicotine. The three different principles of therapeutic action epidemiological findings and large clinical prospective trials reporting a are addressed: active and passive immunotherapy, modulation of alpha-synuclein aggregation, and enhancement of autophagy of alpha-synuclein. -

Table of Contents
PI name: Carsten G. Bönnemann, MD Date: 01-Dec-2017 NIH Protocol number: 15-N-0018 NIH Version: 16 Santhera Protocol number: SNT-I-015 Santhera Version: 10 Congenital Muscular Dystrophy Ascending Multiple Dose Cohort Study anaLyzing Pharmacokinetics at three dose Levels In Children and Adolescents with Assessment of Safety and Tolerability of Omigapil (CALLISTO) Clinical Research Protocol Abbreviated Title: CALLISTO NIH Protocol Number: 15-N-0018 Santhera Protocol Number: SNT-I-015 Date of This Submission/Version (see header) IND Number: 113,370 Eudract Number: 2012-002700-40 Compound: Omigapil Hydrogen Maleate CRADA 2012-0097 (expiration 2020) Confidential Property of Santhera Pharmaceuticals (Switzerland) Ltd. 1 PI name: Carsten G. Bönnemann, MD Date: 01-Dec-2017 NIH Protocol number: 15-N-0018 NIH Version: 16 Santhera Protocol number: SNT-I-015 Santhera Version: 10 Synopsis Study Title Congenital Muscular Dystrophy Ascending Multiple Dose Cohort Study anaLyzing Pharmacokinetics at three dose Levels In Children and Adolescents with Assessment of Safety and Tolerability of Omigapil Study Number SNT-I-015 Study Phase 1 Study Acronym CALLISTO Planned Study Duration 84 weeks (16 weeks recruitment, 68 weeks study conduct). The above duration is assuming cohorts are dosed in staggered parallel. Patient participation will be up to 28 weeks (up to 4 weeks screening, 4 weeks run-in, 12 weeks treatment and a follow-up visit 8 weeks post last dose taken) Investigational Product Omigapil oral solution (0.1 and 0.25 mg/ml in Humco® Cherry Syrup) -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

TREAT-NMD Newsletter No.34
TREATTREAT --NMDNMD NeuromuscularNeuromuscular NetworkNetwork 30th May 2008 Newsletter No. 34 Welcome to the latest newsletter. This edition features Newsletter contents a report from the EuroBioBank Annual Meeting and a call for Eastern European applicants for 6 TREAT-NMD Fellowships to attend the WMS Teaching Course. About this newsletter ……………………………. 1 The TREAT-NMD web site is currently migrating to a Working with us…………………………………… 1 new format and the web pages will be updated on an TREAT-NMD news and reports……………….. 2 ongoing basis over the coming days. Please bear with us Other news………………………………………….. 5 during this transitional period. Thank you. Resources...………….....………………………….. 6 Please forward any items that you would like to be Meetings and workshops…….……………....... 7 included in future editions to [email protected] . Partner section …………..…….……………....... 7 Best wishes, The TREAT-NMD coordination team ***IMPORTANT MESSAGE TO ALL OUR READERS*** We’d like to remind readers that receiving this newsletter does not automatically make you a member of TREAT-NMD. If you have not completed our membership application form, you are not a member! If you are interested in developing closer links with us, we encourage you to read our Members’ Charter and complete the membership application form. These documents can be found on our web site at the address below. Become a Member of TREAT-NMD If you are interested in becoming a member of the TREAT-NMD Network please visit our web site to download our membership charter. An application form is also available for download. The web link to our Members’ sec- tion is: http://www.treat-nmd.eu/research/get-involved/ . -

The Clinical Trial Landscape in Amyotrophic Lateral Sclerosis—Past, Present, and Future
Received: 16 September 2019 | Revised: 8 December 2019 | Accepted: 27 January 2020 DOI: 10.1002/med.21661 REVIEW ARTICLE The clinical trial landscape in amyotrophic lateral sclerosis—Past, present, and future Heike J. Wobst1 | Korrie L. Mack2,3 | Dean G. Brown4 | Nicholas J. Brandon1 | James Shorter2 1Neuroscience, BioPharmaceuticals R&D, AstraZeneca, Boston, Massachusetts Abstract 2 Department of Biochemistry and Biophysics, Amyotrophic lateral sclerosis (ALS) is a fatal neurodegen- Perelman School of Medicine, University of erative disease marked by progressive loss of muscle func- Pennsylvania, Philadelphia, Pennsylvania ‐ 3Merck & Co, Inc, Kenilworth, New Jersey tion. It is the most common adult onset form of motor 4Hit Discovery, Discovery Sciences, neuron disease, affecting about 16 000 people in the United BioPharmaceuticals R&D, AstraZeneca, Boston, States alone. The average survival is about 3 years. Only two Massachusetts interventional drugs, the antiglutamatergic small‐molecule Correspondence riluzole and the more recent antioxidant edaravone, have Heike J. Wobst, Jnana Therapeutics, Northern been approved for the treatment of ALS to date. Therapeutic Avenue, Boston, MA 02210. Email: [email protected] strategies under investigation in clinical trials cover a range of different modalities and targets, and more than 70 dif- James Shorter, Department of Biochemistry and Biophysics, Perelman School of Medicine, ferent drugs have been tested in the clinic to date. Here, we University of Pennsylvania, Philadelphia, PA summarize and classify interventional therapeutic strategies 19104. Email: [email protected] based on their molecular targets and phenotypic effects. We also discuss possible reasons for the failure of clinical trials in Present address Heike J. Wobst, Dean G. -

(12) Patent Application Publication (10) Pub. No.: US 2016/0354335 A1 COHEN Et Al
US 20160354335A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0354335 A1 COHEN et al. (43) Pub. Date: Dec. 8, 2016 (54) COMBINATION OF BACLOFEN, Publication Classification ACAMPROSATE AND MEDIUM CHAN TRGLYCERDES FOR THE TREATMENT (51) Int. Cl. OF NEUROLOGICAL DISORDERS A6II 3L/23 (2006.01) A6II 3/85 (2006.01) (71) Applicant: PHARNEXT, Issy Les Moulineaux A636/85 (2006.01) (FR) A 6LX 3L/97 (2006.01) (52) U.S. Cl. (72) Inventors: DANIEL COHEN, SAINT CLOUD CPC ............. A6 IK3I/23 (2013.01); A61K 31/197 (FR), ILYA CHUMAKOV, (2013.01); A61K 31/185 (2013.01); A61 K VAUX-LE-PENIL (FR), SERGUEI 36/185 (2013.01) NABIROCHKIN, CHATENAY-MALABRY (FR): (57) ABSTRACT RODOLPHE HAJJ, SAINT The present invention relates to combinations and methods GERMAIN EN LAYE (FR) for the treatment of neurological disorders related Amyloid (21) Appl. No.: 15/117,774 beta toxicity and/or neuronal death and/or glucose impaired neuronal metabolism. More specifically, the present inven (22) PCT Fed: Feb. 10, 2015 tion relates to novel combinatorial therapies of Alzheimer's disease, Alzheimer's disease related disorders, frontotempo (86) PCT No.: PCT/EP2015/052694 ral dementia, Parkinson's disease, Lewy body dementia, Huntington's disease, peripheral neuropathies, alcoholism S 371 (c)(1), or alcohol withdrawal, neurological manifestations of drug (2) Date: Aug. 10, 2016 abuse or drug abuse withdrawal, amyotrophic lateral scle rosis, multiple Sclerosis, spinal cord injury, epilepsy, trau Related U.S. Application Data matic brain injury or brain ischemic events based on (60) Provisional application No. 61/938,340, filed on Feb. -

2016 Medicines in Development for Rare Diseases a LIST of ORPHAN DRUGS in the PIPELINE
2016 Medicines in Development for Rare Diseases A LIST OF ORPHAN DRUGS IN THE PIPELINE Autoimmune Diseases Product Name Sponsor Official FDA Designation* Development Status Actemra® Genentech treatment of systemic sclerosis Phase III tocilizumab South San Francisco, CA www.gene.com Adempas® Bayer HealthCare Pharmaceuticals treatment of systemic sclerosis Phase II riociguat Whippany, NJ www.pharma.bayer.com ARA 290 Araim Pharmaceuticals treatment of neuropathic pain in patients Phase II Tarrytown, NY with sarcoidosis www.ariampharma.com ARG201 arGentis Pharmaceuticals treatment of diffuse systemic sclerosis Phase II (type 1 native bovine skin Collierville, TN www.argentisrx.com collagen) BYM338 Novartis Pharmaceuticals treatment of inclusion body myositis Phase III (bimagrumab) East Hanover, NJ www.novartis.com CCX168 ChemoCentryx treatment of anti-neutrophil cytoplasmic Phase II (5a receptor antagonist) Mountain View, CA auto-antibodies associated vasculitides www.chemocentryx.com (granulomatosis with polyangitis or Wegener's granulomatosis), microscopic polyangitis, and Churg-Strauss syndrome * This designation is issued by the FDA's Office of Orphan Products Development while the drug is still in development. The designation makes the sponsor of the drug eligible for entitlements under the Orphan Drug Act of 1983. The entitlements include seven years of marketing exclusivity following FDA approval of the drug for the designated use. Medicines in Development: Rare Diseases | 2016 1 Autoimmune Diseases Product Name Sponsor Official FDA -

Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Frontiers - Publisher Connector REVIEW published: 18 October 2016 doi: 10.3389/fphar.2016.00340 Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology John P. M. Finberg 1* and Jose M. Rabey 2 1 Rappaport Faculty of Medicine, Technion, Israel Institute of Technology, Haifa, Israel, 2 Assaf Harofe Medical Center, Affiliated to Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel Inhibitors of MAO-A and MAO-B are in clinical use for the treatment of psychiatric and neurological disorders respectively. Elucidation of the molecular structure of the active sites of the enzymes has enabled a precise determination of the way in which substrates and inhibitor molecules are metabolized, or inhibit metabolism of substrates, respectively. Despite the knowledge of the strong antidepressant efficacy of irreversible MAO inhibitors, their clinical use has been limited by their side effect of potentiation of the cardiovascular effects of dietary amines (“cheese effect”). A number of reversible MAO-A inhibitors which are devoid of cheese effect have been described in the literature, but only one, moclobemide, is currently in clinical use. The irreversible inhibitors of MAO-B, selegiline and rasagiline, are used clinically in treatment of Parkinson’s disease, and a recently introduced reversible MAO-B inhibitor, safinamide, has also been found efficacious. Modification of the pharmacokinetic characteristics of selegiline by transdermal administration has led to the development of a new drug form for treatment of depression. The clinical potential of MAO inhibitors together with detailed knowledge Edited by: of the enzyme’s binding site structure should lead to future developments with these Rona R. -

P2X7 Receptor Antagonism As a Potential Therapy in Amyotrophic Lateral Sclerosis
REVIEW published: 12 June 2020 doi: 10.3389/fnmol.2020.00093 P2X7 Receptor Antagonism as a Potential Therapy in Amyotrophic Lateral Sclerosis Cristina Ruiz-Ruiz 1†, Francesco Calzaferri 1† and Antonio G. García 1,2* 1Instituto Teófilo Hernando and Departamento de Farmacología, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain, 2Instituto de Investigación Sanitaria, Hospital Universitario de La Princesa, Universidad Autónoma de Madrid, Madrid, Spain This review focuses on the purinergic ionotropic receptor P2X7 (P2X7R) as a potential target for developing drugs that delay the onset and/or disease progression in patients with amyotrophic lateral sclerosis (ALS). Description of clinical and genetic ALS features is followed by an analysis of advantages and drawbacks of transgenic mouse models of disease based on mutations in a bunch of proteins, particularly Cu/Zn superoxide dismutase (SOD1), TAR-DNA binding protein-43 (TDP-43), Fused in Sarcoma/Translocated in Sarcoma (FUS), and Chromosome 9 open reading frame 72 (C9orf72). Though of limited value, these models are however critical to study the proof of concept of new compounds, before reaching clinical trials. The authors also provide a description of ALS pathogenesis including protein aggregation, calcium-dependent excitotoxicity, dysfunction of calcium-binding proteins, ultrastructural mitochondrial Edited by: alterations, disruption of mitochondrial calcium handling, and overproduction of reactive Miguel Diaz-Hernandez, Complutense University of Madrid, oxygen species (ROS). Understanding disease pathogenic pathways may ease the Spain identification of new drug targets. Subsequently, neuroinflammation linked with P2X7Rs Reviewed by: in ALS pathogenesis is described in order to understand the rationale of placing the use Ronald Sluyter, University of Wollongong, Australia of P2X7R antagonists as a new therapeutic pharmacological approach to ALS. -
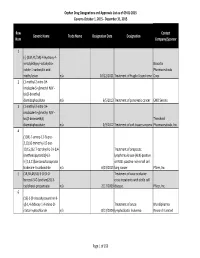
Orphan Drug Designation List
Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 1 (‐)‐(3aR,4S,7aR)‐4‐Hydroxy‐4‐ m‐tolylethynyl‐octahydro‐ Novartis indole‐1‐carboxylic acid Pharmaceuticals methyl ester n/a 10/12/2011 Treatment of Fragile X syndrome Corp. 2 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐broethyl) diamidophosphate n/a 6/5/2013 Treatment of pancreatic cancer EMD Serono 3 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐bromoethyl) Threshold diamidophosphate n/a 3/9/2012 Treatment of soft tissue sarcoma Pharmaceuticals, Inc. 4 (1OR)‐7‐amino‐12‐fluoro‐ 2,10,16‐trimethyl‐15 oxo‐ 10,15,16,17‐tetrahydro‐2H‐8,4‐ Treatment of anaplastic (metheno)pyrazolo[4,3‐ lymphoma kinase (ALK)‐positive h][2,5,11]benzoxadiazacyclote or ROS1‐positive non‐small cell tradecine‐3‐carbonitrile n/a 6/23/2015 lung cancer Pfizer, Inc. 5 (1R,3R,4R,5S)‐3‐O‐[2‐O‐ Treatment of vaso‐occlusive benzoyl‐3‐O‐(sodium(2S)‐3‐ crisis in patients with sickle cell cyclohexyl‐propanoate‐ n/a 2/17/2009 disease. Pfizer, Inc. 6 (1S)‐1‐(9‐deazahypoxanthin‐9‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ Treatment of acute Mundipharma ribitol‐hydrochloride n/a 8/13/2004 lymphoblastic leukemia Research Limited Page 1 of 359 Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 7 Treatment of chronic lymphocytic leukemia and related leukemias to include (1S)‐1‐(9‐deazahypoxanthin‐9‐ prolymphocytic leukemia, adult T‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ cell leukemia, and hairy cell Mundipharma ribitol‐hydrochloride n/a 8/10/2004 leukemia Research Ltd. -

Neurological Disorders and the Blood-Brain Barrier
Page 362 to 383 Volume 2 • Issue 1 • 2018 Review Article Current Opinions in Neurological Science ISSN: 2575-5447 Neurological Disorders and the Blood Brain Barrier: 2. Parkinson and Other Movement Disorders Alain L Fymat* International Institute of Medicine and Science, California, USA *Corresponding Author: Alain L Fymat, International Institute of Medicine and Science, California, USA. Received: January 10, 2018; Published: January 25, 2018 Abstract While the extent of our body of knowledge of Parkinson’s disease (PD) and other movement disorders and the number of cor- responding treatment drugs are both vast, there is still no cure for these chronic and relentlessly progressive diseases. The motor symptoms of PD are known to result from reduced dopamine production in the brain’s basal ganglia. However, Dopamine does not cross the blood brain barrier (BBB) so it cannot be taken as a medicine to boost the brain’s depleted levels of Dopamine. However a precursor of Dopamine, Levodopa, can pass through this barrier to the brain where it is readily converted to Dopamine. Administra- tion of this drug temporarily diminishes the motor symptoms of PD. Unfortunately, only 5–10% of the drug crosses the barrier with - cuss the current treatments of motor symptoms prior to treatment with the mainstay L-dopa-based therapy and later as the disease much of the remainder being metabolized to Dopamine elsewhere in the body, where it causes a variety of side effects. I will first dis progresses and other pharmacologic treatment options as well as the numerous pharmacotherapies for non-motor symptoms. For early to very advanced PD patients, I will also discuss deep brain stimulation (DBS), which leads to less motor or neuropsychiatric adverse effects, or both.