Proefschrift Maurice Van Staveren.Indb
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metabolism of Purines and Pyrimidines in Health and Disease
39th Meeting of the Polish Biochemical Society Gdañsk 16–20 September 2003 SESSION 6 Metabolism of purines and pyrimidines in health and disease Organized by A. C. Sk³adanowski, A. Guranowski 182 Session 6. Metabolism of purines and pyrimidines in health and disease 2003 323 Lecture The role of DNA methylation in cytotoxicity mechanism of adenosine analogues in treatment of leukemia Krystyna Fabianowska-Majewska Zak³ad Chemii Medycznej IFiB, Uniwersytet Medyczny, ul. Mazowiecka 6/8, 92 215 £ódŸ Changes in DNA methylation have been recognized tory effects of cladribine and fludarabine on DNA as one of the most common molecular alterations in hu- methylation, after 48 hr growth of K562 cells with the man neoplastic diseases and hypermethylation of drugs, are non-random and affect mainly CpG rich is- gene-promoter regions is one of the most frequent lands or CCGG sequences but do not affect sepa- mechanisms of the loss of gene functions. For this rea- rately-located CpG sequences. The analysis showed son, DNA methylation may be a tool for detection of that cladribine (0.1 mM) reduced the methylated early cell transformations as well as predisposition to cytosines in CpG islands and CCGG sequences to a sim- metastasis process. Moreover, DNA methylation seems ilar degree. The inhibition of cytosine methylation by to be a promissing target for new preventive and thera- fludarabine (3 mM) was observed mainly in CCGG se- peutic strategies. quences, sensitive to HpaII, but the decline in the meth- Our studies on DNA methylation and cytotoxicity ylated cytosine, located in CpG island was 2-fold lower mechanism of antileukemic drugs, cladribine and than that with cladribine. -

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -

Transcriptomic and Proteomic Profiling Provides Insight Into
BASIC RESEARCH www.jasn.org Transcriptomic and Proteomic Profiling Provides Insight into Mesangial Cell Function in IgA Nephropathy † † ‡ Peidi Liu,* Emelie Lassén,* Viji Nair, Celine C. Berthier, Miyuki Suguro, Carina Sihlbom,§ † | † Matthias Kretzler, Christer Betsholtz, ¶ Börje Haraldsson,* Wenjun Ju, Kerstin Ebefors,* and Jenny Nyström* *Department of Physiology, Institute of Neuroscience and Physiology, §Proteomics Core Facility at University of Gothenburg, University of Gothenburg, Gothenburg, Sweden; †Division of Nephrology, Department of Internal Medicine and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan; ‡Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya, Japan; |Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden; and ¶Integrated Cardio Metabolic Centre, Karolinska Institutet Novum, Huddinge, Sweden ABSTRACT IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes. -

Dihydropyrimidinase Deficiency
Dihydropyrimidinase deficiency Description Dihydropyrimidinase deficiency is a disorder that can cause neurological and gastrointestinal problems in some affected individuals. Other people with dihydropyrimidinase deficiency have no signs or symptoms related to the disorder, and in these individuals the condition can be diagnosed only by laboratory testing. The neurological abnormalities that occur most often in people with dihydropyrimidinase deficiency are intellectual disability, seizures, and weak muscle tone (hypotonia). An abnormally small head size (microcephaly) and autistic behaviors that affect communication and social interaction also occur in some individuals with this condition. Gastrointestinal problems that occur in dihydropyrimidinase deficiency include backflow of acidic stomach contents into the esophagus (gastroesophageal reflux) and recurrent episodes of vomiting (cyclic vomiting). Affected individuals can also have deterioration ( atrophy) of the small, finger-like projections (villi) that line the small intestine and provide a large surface area with which to absorb nutrients. This condition, called villous atrophy, can lead to difficulty absorbing nutrients from foods (malabsorption), resulting in a failure to grow and gain weight at the expected rate (failure to thrive). People with dihydropyrimidinase deficiency, including those who otherwise exhibit no symptoms, may be vulnerable to severe, potentially life-threatening toxic reactions to certain drugs called fluoropyrimidines that are used to treat cancer. Common examples of these drugs are 5-fluorouracil and capecitabine. These drugs may not be broken down efficiently and can build up to toxic levels in the body (fluoropyrimidine toxicity), leading to drug reactions including gastrointestinal problems, blood abnormalities, and other signs and symptoms. Frequency Dihydropyrimidinase deficiency is thought to be a rare disorder. -

1611 REGULATION of PYRIMIDINE METABOLISM in PLANTS Chris
[Frontiers in Bioscience 9, 1611-1625, May 1, 2004] REGULATION OF PYRIMIDINE METABOLISM IN PLANTS 1, 2 1, 3 1, 4 1, 5 1, 6 1, 7 Chris Kafer , Lan Zhou , Djoko Santoso , Adel Guirgis , Brock Weers , Sanggyu Park and Robert Thornburg 1 1 Department of Biochemistry, Biophysics, and Molecular Biology, Iowa State University, Ames, Iowa 50011, 2 BASF Plant Science LLC, 2901 South Loop Drive, Ste 3800, Ames, Iowa 50014, 3 Lan Zhou, Pioneer Hi-Bred International, Inc. 7300 NW 62nd Avenue, PO Box 1004, Johnston, Iowa 50131-1004, 4 Indonesian Biotechnology Research Institute for Estate Crops, Jl, Taman Kencana No 1, Bogor 16151 Indonesia, 5 Institute of Genetic Engineering and Biotechnology, Menofiya University, PO Box 79/22857, Sadat City, Egypt, 6 Department of Biochemistry, University of Iowa, 4/511 Bowen Science Building, Iowa City, Iowa 52242-1109, 7 Division of Life and Environment, College of Natural Resources, Daegu University, Gyongsan City, Gyongbuk, Korea 712-714 TABLE OF CONTENTS 1. Abstract 2. Introduction 3. Pyrimidine metabolic pathways 3.1. De novo pyrimidine biosynthesis 3.1.1. CPSase 3.1.2. ATCase 3.1.3. DHOase 3.1.4. DHODH 3.1.5. UMPS 3.1.6. Intracellular Organization of the de novo Pathway 3.2. Pyrimidine Salvage and Recycling 3.2.1. Cytosine deaminase 3.2.2. Cytidine deaminase 3.2.3. UPRTase 3.3. Pyrimidine Modification 3.3.1. UMP/CMP kinase 3.3.2. NDP kinase 3.3.3. CTP synthase, NDP reductase, dUTPase 3.3.4. Thymidylate synthase/Dihydrofolate reductase 3.4. Pyrimidine Catabolism 4. Regulation of pyrimidine metabolism 4.1. -

Regulation of the Glutamate/Glutamine Cycle by Nitric Oxide in the Central Nervous System
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2015 Regulation of the Glutamate/glutamine Cycle by Nitric Oxide in the Central Nervous System Karthik Anderson Raju University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons, Biology Commons, and the Neuroscience and Neurobiology Commons Recommended Citation Raju, Karthik Anderson, "Regulation of the Glutamate/glutamine Cycle by Nitric Oxide in the Central Nervous System" (2015). Publicly Accessible Penn Dissertations. 1962. https://repository.upenn.edu/edissertations/1962 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1962 For more information, please contact [email protected]. Regulation of the Glutamate/glutamine Cycle by Nitric Oxide in the Central Nervous System Abstract Nitric oxide (˙NO) is a critical contributor to glutamatergic neurotransmission in the central nervous system (CNS). Much of its influence is due ot the ability of this molecule to regulate protein structure and function through its posttranslational modification of cysteine esidues,r a process known as S- nitrosylation. However, little is known about the extent of this modification and its associated functional effects in the brain under physiological conditions. We employed mass spectrometry (MS)-based methodologies to interrogate the S-nitrosocysteine proteome in wild-type (WT), neuronal nitric oxide synthase-deficient (nNOS-/-), -

Dihydropyrimidine Dehydrogenase in the Metabolism of the Anticancer Drugs
Cancer Chemotherapy and Pharmacology (2019) 84:1157–1166 https://doi.org/10.1007/s00280-019-03936-w REVIEW ARTICLE Dihydropyrimidine dehydrogenase in the metabolism of the anticancer drugs Vinay Sharma1 · Sonu Kumar Gupta1 · Malkhey Verma1 Received: 3 May 2019 / Accepted: 21 August 2019 / Published online: 4 September 2019 © Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract Cancer caused by fundamental defects in cell cycle regulation leads to uncontrolled growth of cells. In spite of the treatment with chemotherapeutic agents of varying nature, multiple resistance mechanisms are identifed in cancer cells. Similarly, numerous variations, which decrease the metabolism of chemotherapeutics agents and thereby increasing the toxicity of anticancer drugs have been identifed. 5-Fluorouracil (5-FU) is an anticancer drug widely used to treat many cancers in the human body. Its broad targeting range is based upon its capacity to act as a uracil analogue, thereby disrupting RNA and DNA synthesis. Dihydropyrimidine dehydrogenase (DPD) is an enzyme majorly involved in the metabolism of pyrimidines in the human body and has the same metabolising efect on 5-FU, a pyrimidine analogue. Multiple mutations in the DPD gene have been linked to 5-FU toxicity and inadequate dosages. DPD inhibitors have also been used to inhibit excessive degradation of 5-FU for meeting appropriate dosage requirements. This article focusses on the role of dihydropyrimidine dehydrogenase in the metabolism of the anticancer drug 5-FU and other associated drugs. Keywords Cancer · Anticancer drugs · Dihydropyrimidine dehydrogenase (DPD) · 5-Fluorouracil (5-FU) · Drug resistance · Drug metabolism Introduction by Dihydropyrimidine dehydrogenase (DPD) through the pyrimidine degradation pathway. -
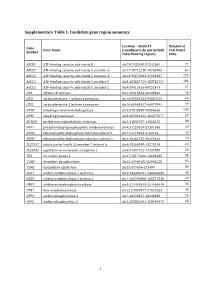
Supplementary Table 1. Candidate Gene Region Summary
Supplementary Table 1. Candidate gene region summary Location – Build 37 Number of Gene Gene Name (coordinates do not include Test Panel Symbol 25kb flanking region) SNPs ABCB1 ATP-binding cassette, sub-family B chr7:87132948-87342564 77 ABCC3 ATP-binding cassette, sub-family C, member 3 chr17:48712218-48769063 64 ABCC4 ATP-binding cassette, sub-family C, member 4 chr13:95672083-95953687 224 ABCC5 ATP-binding cassette, sub-family C, member 5 chr3:183637724-183735727 101 ABCG2 ATP-binding cassette, sub-family G, member 2 chr4:89011416-89152474 57 CDA cytidine deaminase chr1:20915444-20945400 26 CES1 carboxylesterase 1 isoform a precursor chr16:55836764-55867075 24 CES2 carboxylesterase 2 isoform a precursor chr16:66968347-66978994 59 DPYD dihydropyrimidine dehydrogenase chr1:97543300-98386615 239 DPYS dihydropyrimidinase chr8:105391652-105479277 69 MTHFR methylenetetrahydrofolate reductase chr1:11845787-11866115 38 PPAT phosphoribosyl pyrophosphate amidotransferase chr4:57259529-57301845 29 RRM1 ribonucleoside-diphosphate reductase subunit 1 chr11:4115924-4160106 29 RRM2 ribonucleoside-diphosphate reductase subunit 2 chr2:10262735-10270623 19 SLC22A7 solute carrier family 22 member 7 isoform b chr6:43265998-43273276 26 SLC29A1 equilibrative nucleoside transporter 1 chr6:44187242-44201888 26 TK1 thymidine kinase 1 chr17:76170160-76183285 35 TYMP thymidine phosphorylase chr22:50964182-50968258 92 TYMS thymidylate synthetase chr18:657604-673499 34 UCK1 uridine-cytidine kinase 1 isoform a chr9:134399191-134406655 43 UCK2 uridine-cytidine kinase 2 isoform a chr1:165796890-165877339 22 UMPS uridine monophosphate synthase chr3:124449213-124464040 34 UPB1 beta-ureidopropionase chr22:24890077-24922553 30 UPP1 uridine phosphorylase 1 chr7:48128355-48148330 16 UPP2 uridine phosphorylase 2 chr2:158851691-158992478 43 1 Supplementary Table 2. -

Genetic Factors Influencing Pyrimidine- Antagonist Chemotherapy
The Pharmacogenomics Journal (2005) 5, 226–243 & 2005 Nature Publishing Group All rights reserved 1470-269X/05 $30.00 www.nature.com/tpj REVIEW Genetic factors influencing Pyrimidine- antagonist chemotherapy JG Maring1 ABSTRACT 2 Pyrimidine antagonists, for example, 5-fluorouracil (5-FU), cytarabine (ara-C) HJM Groen and gemcitabine (dFdC), are widely used in chemotherapy regimes for 2 FM Wachters colorectal, breast, head and neck, non-small-cell lung cancer, pancreatic DRA Uges3 cancer and leukaemias. Extensive metabolism is a prerequisite for conversion EGE de Vries4 of these pyrimidine prodrugs into active compounds. Interindividual variation in the activity of metabolising enzymes can affect the extent of 1Department of Pharmacy, Diaconessen Hospital prodrug activation and, as a result, act on the efficacy of chemotherapy Meppel & Bethesda Hospital Hoogeveen, Meppel, treatment. Genetic factors at least partly explain interindividual variation in 2 The Netherlands; Department of Pulmonary antitumour efficacy and toxicity of pyrimidine antagonists. In this review, Diseases, University of Groningen & University Medical Center Groningen, Groningen, The proteins relevant for the efficacy and toxicity of pyrimidine antagonists will Netherlands; 3Department of Pharmacy, be summarised. In addition, the role of germline polymorphisms, tumour- University of Groningen & University Medical specific somatic mutations and protein expression levels in the metabolic Center Groningen, Groningen, The Netherlands; pathways and clinical pharmacology -

Metabolic Network-Based Stratification of Hepatocellular Carcinoma Reveals Three Distinct Tumor Subtypes
Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes Gholamreza Bidkhoria,b,1, Rui Benfeitasa,1, Martina Klevstigc,d, Cheng Zhanga, Jens Nielsene, Mathias Uhlena, Jan Borenc,d, and Adil Mardinoglua,b,e,2 aScience for Life Laboratory, KTH Royal Institute of Technology, SE-17121 Stockholm, Sweden; bCentre for Host-Microbiome Interactions, Dental Institute, King’s College London, SE1 9RT London, United Kingdom; cDepartment of Molecular and Clinical Medicine, University of Gothenburg, SE-41345 Gothenburg, Sweden; dThe Wallenberg Laboratory, Sahlgrenska University Hospital, SE-41345 Gothenburg, Sweden; and eDepartment of Biology and Biological Engineering, Chalmers University of Technology, SE-41296 Gothenburg, Sweden Edited by Sang Yup Lee, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea, and approved November 1, 2018 (received for review April 27, 2018) Hepatocellular carcinoma (HCC) is one of the most frequent forms of of markers associated with recurrence and poor prognosis (13–15). liver cancer, and effective treatment methods are limited due to Moreover, genome-scale metabolic models (GEMs), collections tumor heterogeneity. There is a great need for comprehensive of biochemical reactions, and associated enzymes and transporters approaches to stratify HCC patients, gain biological insights into have been successfully used to characterize the metabolism of subtypes, and ultimately identify effective therapeutic targets. We HCC, as well as identify drug targets for HCC patients (11, 16–18). stratified HCC patients and characterized each subtype using tran- For instance, HCC tumors have been stratified based on the uti- scriptomics data, genome-scale metabolic networks and network lization of acetate (11). Analysis of HCC metabolism has also led topology/controllability analysis. -

Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes
JASN Express. Published on December 3, 2008 as doi: 10.1681/ASN.2008040406 BASIC RESEARCH www.jasn.org Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes Patricia A. Gonzales,*† Trairak Pisitkun,* Jason D. Hoffert,* Dmitry Tchapyjnikov,* ʈ Robert A. Star,‡ Robert Kleta,§ ¶ Nam Sun Wang,† and Mark A. Knepper* *Laboratory of Kidney and Electrolyte Metabolism, National Heart, Lung, and Blood Institute, ‡Renal Diagnostics and Therapeutics Unit, National Institute of Diabetes and Digestive and Kidney Diseases, §Section of Human ʈ Biochemical Genetics, Medical Genetics Branch, National Human Genome Research Institute, and Office of Rare Diseases, Office of the Director, National Institutes of Health, Bethesda, and †Department of Chemical and Biomolecular Engineering, University of Maryland, College Park, Maryland; and ¶London Epithelial Group, Centre for Nephrology, University College London, London, United Kingdom ABSTRACT Normal human urine contains large numbers of exosomes, which are 40- to 100-nm vesicles that originate as the internal vesicles in multivesicular bodies from every renal epithelial cell type facing the urinary space. Here, we used LC-MS/MS to profile the proteome of human urinary exosomes. Overall, the analysis identified 1132 proteins unambiguously, including 177 that are represented on the Online Mendelian Inheritance in Man database of disease-related genes, suggesting that exosome analysis is a potential approach to discover urinary biomarkers. We extended the proteomic analysis to phospho- proteomic profiling using neutral loss scanning, and this yielded multiple novel phosphorylation sites, including serine-811 in the thiazide-sensitive Na-Cl co-transporter, NCC. To demonstrate the potential use of exosome analysis to identify a genetic renal disease, we carried out immunoblotting of exosomes from urine samples of patients with a clinical diagnosis of Bartter syndrome type I, showing an absence of the sodium-potassium-chloride co-transporter 2, NKCC2. -

Genetic Polymorphisms of Dihydropyrimidinase in a Japanese Patient with Capecitabine-Induced Toxicity
RESEARCH ARTICLE Genetic Polymorphisms of Dihydropyrimidinase in a Japanese Patient with Capecitabine-Induced Toxicity Masahiro Hiratsuka1☯*, Hiroshi Yamashita2☯, Fumika Akai1, Hiroki Hosono1, Eiji Hishinuma1, Noriyasu Hirasawa1, Takahiro Mori3☯ 1 Laboratory of Pharmacotherapy of Life-Style Related Diseases, Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan, 2 Department of Surgery, Iwate Prefectural Chubu Hospital, Kitakami, Japan, 3 Graduate School of Medicine, Tohoku University, Sendai, Japan ☯ These authors contributed equally to this work. * [email protected] Abstract Dihydropyrimidinase (DHP) is the second enzyme in the catabolic pathway of uracil, thy- mine, and chemotherapeutic fluoropyrimidine agents such as 5-fluorouracil (5-FU). Thus, DHP deficiency might be associated with 5-FU toxicity during fluoropyrimidine chemothera- py. We performed genetic analyses of the family of a patient with advanced colon cancer OPEN ACCESS who underwent radical colectomy followed by treatment with 5-FU prodrug capecitabine and developed severe toxicity attributable to a lack of DHP. We measured urinary uracil and Citation: Hiratsuka M, Yamashita H, Akai F, Hosono H, Hishinuma E, Hirasawa N, et al. (2015) Genetic dihydrouracil, and genotyped DPYS in the patient and her family. We also measured the Polymorphisms of Dihydropyrimidinase in a allele frequency of DPYS polymorphisms in 391 unrelated Japanese subjects. The patient Japanese Patient with Capecitabine-Induced Toxicity. had compound heterozygous missense and nonsense polymorphisms comprising PLoS ONE 10(4): e0124818. doi:10.1371/journal. > > pone.0124818 c.1001A G (p.Gln334Arg) in exon 6 and c.1393C T (p.Arg465Ter) in exon 8, which are known to result in a DHP enzyme with little or no activity.