P53-Dependent Inhibition of FKHRL1 in Response to DNA Damage Through Protein Kinase SGK1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Celastrol Suppresses Experimental Autoimmune Encephalomyelitis Via MAPK/SGK1-Regulated Mediators of Autoimmune Pathology
Inflammation Research (2019) 68:285–296 https://doi.org/10.1007/s00011-019-01219-x Inflammation Research ORIGINAL RESEARCH PAPER Celastrol suppresses experimental autoimmune encephalomyelitis via MAPK/SGK1-regulated mediators of autoimmune pathology Shivaprasad H. Venkatesha1,2 · Kamal D. Moudgil1,2,3 Received: 17 November 2018 / Revised: 10 January 2019 / Accepted: 11 February 2019 / Published online: 28 February 2019 © This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply 2019 Abstract Objective and design Multiple sclerosis (MS) is a debilitating autoimmune disease involving immune dysregulation of the pathogenic T helper 17 (Th17) versus protective T regulatory (Treg) cell subsets, besides other cellular aberrations. Studies on the mechanisms underlying these changes have unraveled the involvement of mitogen-activated protein kinase (MAPK) pathway in the disease process. We describe here a gene expression- and bioinformatics-based study showing that celastrol, a natural triterpenoid, acting via MAPK pathway regulates the downstream genes encoding serum/glucocorticoid regulated kinase 1 (SGK1), which plays a vital role in Th17/Treg differentiation, and brain-derived neurotrophic factor (BDNF), which is a neurotrophic factor, thereby offering protection against experimental autoimmune encephalomyelitis (EAE) in mice. Methods We first tested the gene expression profile of splenocytes of EAE mice in response to the disease-related antigen, myelin oligodendrocyte glycoprotein (MOG), and then examined the effect of celastrol on that profile. Results Interestingly, celastrol reversed the expression of many MOG-induced genes involved in inflammation and immune pathology. The MAPK pathway involving p38MAPK and ERK was identified as one of the mediators of celastrol action. -

The Expression of Genes Contributing to Pancreatic Adenocarcinoma Progression Is Influenced by the Respective Environment – Sagini Et Al
The expression of genes contributing to pancreatic adenocarcinoma progression is influenced by the respective environment – Sagini et al Supplementary Figure 1: Target genes regulated by TGM2. Figure represents 24 genes regulated by TGM2, which were obtained from Ingenuity Pathway Analysis. As indicated, 9 genes (marked red) are down-regulated by TGM2. On the contrary, 15 genes (marked red) are up-regulated by TGM2. Supplementary Table 1: Functional annotations of genes from Suit2-007 cells growing in pancreatic environment Categoriesa Diseases or p-Valuec Predicted Activation Number of genesf Functions activationd Z-scoree Annotationb Cell movement Cell movement 1,56E-11 increased 2,199 LAMB3, CEACAM6, CCL20, AGR2, MUC1, CXCL1, LAMA3, LCN2, COL17A1, CXCL8, AIF1, MMP7, CEMIP, JUP, SOD2, S100A4, PDGFA, NDRG1, SGK1, IGFBP3, DDR1, IL1A, CDKN1A, NREP, SEMA3E SERPINA3, SDC4, ALPP, CX3CL1, NFKBIA, ANXA3, CDH1, CDCP1, CRYAB, TUBB2B, FOXQ1, SLPI, F3, GRINA, ITGA2, ARPIN/C15orf38- AP3S2, SPTLC1, IL10, TSC22D3, LAMC2, TCAF1, CDH3, MX1, LEP, ZC3H12A, PMP22, IL32, FAM83H, EFNA1, PATJ, CEBPB, SERPINA5, PTK6, EPHB6, JUND, TNFSF14, ERBB3, TNFRSF25, FCAR, CXCL16, HLA-A, CEACAM1, FAT1, AHR, CSF2RA, CLDN7, MAPK13, FERMT1, TCAF2, MST1R, CD99, PTP4A2, PHLDA1, DEFB1, RHOB, TNFSF15, CD44, CSF2, SERPINB5, TGM2, SRC, ITGA6, TNC, HNRNPA2B1, RHOD, SKI, KISS1, TACSTD2, GNAI2, CXCL2, NFKB2, TAGLN2, TNF, CD74, PTPRK, STAT3, ARHGAP21, VEGFA, MYH9, SAA1, F11R, PDCD4, IQGAP1, DCN, MAPK8IP3, STC1, ADAM15, LTBP2, HOOK1, CST3, EPHA1, TIMP2, LPAR2, CORO1A, CLDN3, MYO1C, -

Region-Specific Transcriptional Changes Following the Three
Molecular Psychiatry (2007) 12, 167–189 & 2007 Nature Publishing Group All rights reserved 1359-4184/07 $30.00 www.nature.com/mp ORIGINAL ARTICLE Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine B Conti1, R Maier2, AM Barr1,3, MC Morale1,4,XLu1, PP Sanna1, G Bilbe2, D Hoyer2 and T Bartfai1 1Molecular and Integrative Neuroscience Department, The Harold L Dorris Neurological Research Institute, The Scripps Research Institute, La Jolla, CA, USA and 2Neuroscience Research, Novartis Institutes for Biomedical Research, Basel, Switzerland The significant proportion of depressed patients that are resistant to monoaminergic drug therapy and the slow onset of therapeutic effects of the selective serotonin reuptake inhibitors (SSRIs)/serotonin/noradrenaline reuptake inhibitors (SNRIs) are two major reasons for the sustained search for new antidepressants. In an attempt to identify common underlying mechanisms for fast- and slow-acting antidepressant modalities, we have examined the transcriptional changes in seven different brain regions of the rat brain induced by three clinically effective antidepressant treatments: electro convulsive therapy (ECT), sleep deprivation (SD), and fluoxetine (FLX), the most commonly used slow-onset antidepressant. Each of these antidepressant treatments was applied with the same regimen known to have clinical efficacy: 2 days of ECT (four sessions per day), 24 h of SD, and 14 days of daily treatment of FLX, respectively. Transcriptional changes were evaluated on RNA extracted from seven different brain regions using the Affymetrix rat genome microarray 230 2.0. The gene chip data were validated using in situ hybridization or autoradiography for selected genes. -

Mechanisms of Type I and Type II Pseudohypoaldosteronism
SCIENCE IN RENAL MEDICINE www.jasn.org Mechanisms of Type I and Type II Pseudohypoaldosteronism Seth B. Furgeson and Stuart Linas Division of Renal Diseases and Hypertension, Department of Medicine, University of Colorado, Aurora, Colorado ABSTRACT Pseudohypoaldosteronism (PHA) types I and II are curious genetic disorders that 100 cases with a similar syndrome (vari- share hyperkalemia as a predominant finding. Together they have become win- able levels of salt wasting, metabolic aci- dows to understanding new molecular physiology in the kidney. Autosomal reces- dosis, and hyperkalemia) have since been sive PHAI results from mutations in the epithelial sodium channel (ENaC), whereas described with both autosomal dominant autosomal dominant PHAI is characterized by mutations in the mineralocorticoid and recessive patterns of inheritance. In receptor. PHAII is the result of mutations in a family of serine-threonine kinases 1996, Chang et al.2 reported that autoso- called with-no-lysine kinases (WNK)1 and WNK4. WNK4 negatively regulates the mal recessive PHAI was caused by muta- NaCl cotransporter (NCC), and PHAII mutations in WNK4 abrogate this affect. tions of ENaC. Most cases of autosomal WNK4 also regulates the expression or function of renal outer medullary potassium dominant PHAI link to mutations in the (ROMK) channels, ENaCs, and Cl transporters. WNK1 also regulates NCC and mineralocorticoid receptor (MR).3 In gen- ROMK. Aldosterone inactivates WNK1 and WNK4 activity. Whether angiotensin II eral, patients with autosomal recessive can fine tune the actions of aldosterone is still unclear. PHAI, as opposed to patients with autoso- mal dominant PHAI, have a more severe J Am Soc Nephrol 21: 1842–1845, 2010. -

Inactivation of Corticotropin- Releasing Hormone–Induced Insulinotropic Role by High- Altitude Hypoxia
Diabetes Volume 64, March 2015 785 Ke Hao,1 Fan-Ping Kong,1 Yu-Qi Gao,2 Jia-Wei Tang,1 Jian Chen,2 A. Mark Evans,3 Stafford L. Lightman,4 Xue-Qun Chen,1 and Ji-Zeng Du1 Inactivation of Corticotropin- Releasing Hormone–Induced Insulinotropic Role by High- Altitude Hypoxia Diabetes 2015;64:785–795 | DOI: 10.2337/db14-0500 We have shown that hypoxia reduces plasma insulin, dysfunction and illness, particularly acute mountain sick- which correlates with corticotropin-releasing hormone ness (AMS) (1). During the construction of the Qinghai- (CRH) receptor 1 (CRHR1) in rats, but the mechanism Tibet railway (at altitudes of 3,000–5,000 m) in China, remains unclear. Here, we report that hypobaric hypoxia at .100,000 construction workers were involved, and 51% an altitude of 5,000 m for 8 h enhances rat plasma CRH, of them developed AMS (2). Moreover, since the railway METABOLISM corticosterone, and glucose levels, whereas the plasma began service, .10 million travelers have visited the Tibet insulin and pancreatic ATP/ADP ratio is reduced. In islets region in 2012, of whom 31% developed AMS despite cultured under normoxia, CRH stimulated insulin release in traveling with an oxygen supply on the train (3). Increas- a glucose- and CRH-level–dependent manner by activat- ing CRHR1 and thus the cAMP-dependent protein kinase ing evidence in both humans and animals suggests that pathway and calcium influx through L-type channels. In exposure to either high-altitude or hypobaric hypoxia fl islets cultured under hypoxia, however, the insulinotropic in uences plasma insulin levels and glucose homeostasis, effect of CRH was inactivated due to reduced ATP and depending on the oxygen level and duration of exposure cAMP and coincident loss of intracellular calcium oscilla- (4–9). -
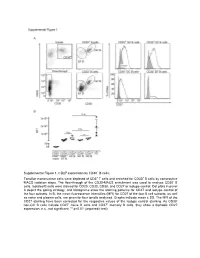
Supplemental Figure 1. CD27 Expression by CD30+ B Cells
Supplemental Figure 1. CD27 expression by CD30+ B cells. Tonsillar mononuclear cells were depleted of CD3+ T cells and enriched for CD30+ B cells by consecutive MACS isolation steps. The flow-through of the CD30-MACS enrichment was used to analyze CD30- B cells. Isolated B cells were stained for CD20, CD30, CD38, and CD27 or isotype control. Dot plots in panel A depict the gating strategy, and histograms show the staining patterns for CD27 and isotype control of the four subsets. In B, the mean fluorescence intensities (MFI) for CD27 of the four B cell subsets, as well as naive and plasma cells, are given for four tonsils analyzed. Graphs indicate mean ± SD. The MFI of the CD27 staining have been corrected for the respective values of the isotype control staining. As CD30- non-GC B cells include CD27- naïve B cells and CD27+ memory B cells, they show a biphasic CD27 expression. n.s., not significant; ** p<0.01 (unpaired t test). Supplemental Figure 2. Interleukin receptor expression by CD30+ B cells and relatedness of CD30+ EF B cells to CD21low B cells. mRNA and surface protein expression of IL2RB (A), IL21R (B) and CD21 (C) are shown for indicated B cell subsets of five tonsils (conventional (conv.) CD30- GC (germinal center) B cells (CD20highCD38+), CD30+ GC B cells, CD30- memory and CD30+ EF (extra follicular) B cells (CD20+CD38-/lowCD27+), and plasma cells (PC) (CD20+CD38high). mRNA data originate from Affymetrix genechip analyses (Tiacci et al., Blood, 2012; 120:4609-4620) and were analyzed using an unpaired t test. -

Serum/Glucocorticoid-Induced Protein Kinase-1 Facilitates Androgen Receptor-Dependent Cell Survival
Cell Death and Differentiation (2007) 14, 2085–2094 & 2007 Nature Publishing Group All rights reserved 1350-9047/07 $30.00 www.nature.com/cdd Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival I Shanmugam1, G Cheng1, PF Terranova2,3, JB Thrasher1,3, CP Thomas4 and B Li*1,2,3 Androgen receptor (AR) is a critical factor in the development and progression of prostate cancer. We and others recently demonstrated that eliminating AR expression leads to apoptotic cell death in AR-positive prostate cancer cells. To understand the mechanisms of AR-dependent survival, we performed a genome-wide search for AR-regulated survival genes. We found that serum/glucocorticoid-induced protein kinase-1 (SGK-1) mRNA levels were significantly upregulated after androgen stimulation, which was confirmed to be AR dependent. Promoter analysis revealed that the AR interacted with the proximal and distal regions of the sgk1 promoter, leading to sgk-1 promoter activation after androgen stimulation. Functional assays demonstrated that SGK-1 was indispensable for the protective effect of androgens on cell death induced by serum starvation. SGK-1 overexpression not only rescued cells from AR small-interfering RNA (siRNA)-induced apoptosis, but also enhanced AR transactivation, even in the absence of androgen. Additionally, SGK-1 siRNA reduced AR transactivation, indicating a positive feedback effect of SGK-1 expression on AR-mediated gene expression and cellular survival. Taken together, our data suggest that SGK-1 is an androgen-regulated gene that plays a pivotal role in AR-dependent survival and gene expression. Cell Death and Differentiation (2007) 14, 2085–2094; doi:10.1038/sj.cdd.4402227; published online 12 October 2007 Androgens play a critical role not only in the physiological Serum and glucocorticoid-induced protein kinase-1 (SGK- development of the prostate but also in the genesis of prostate 1) belongs to the ‘AGC’ subfamily of protein kinases, including cancer (reviewed by Heinlein and Chang1). -

3-Phosphoinositide–Dependent Kinase 1 Potentiates Upstream Lesions on the Phosphatidylinositol 3-Kinase Pathway in Breast Carcinoma
Published OnlineFirst July 14, 2009; DOI: 10.1158/0008-5472.CAN-09-0820 Molecular Biology, Pathobiology, and Genetics 3-Phosphoinositide–Dependent Kinase 1 Potentiates Upstream Lesions on the Phosphatidylinositol 3-Kinase Pathway in Breast Carcinoma Matthew Maurer,1,3 Tao Su,3 Lao H. Saal,4 Susan Koujak,4 Benjamin D. Hopkins,4 Christina R. Barkley,6,7 Jiaping Wu,6 Subhadra Nandula,2 Bhaskar Dutta,9 Yuli Xie,1 Y. Rebecca Chin,8 Da-In Kim,4 Jennifer S. Ferris,5 Sofia K. Gruvberger-Saal,4 Mervi Laakso,10,11 Xiaomei Wang,3 Lorenzo Memeo,12 Albert Rojtman,2 Tulio Matos,2 Jennifer S. Yu,2,4 Carlos Cordon-Cardo,2,3 Jorma Isola,11 MaryBeth Terry, 5 Alex Toker,8 Gordon B. Mills,9 Jean J. Zhao,6 Vundavalli V.V.S. Murty,2,3 Hanina Hibshoosh,2,3 and Ramon Parsons1,2,3,4 Departments of 1Medicine and 2Pathology, 3Herbert Irving Comprehensive Cancer Center, 4Institute for Cancer Genetics, College of Physicians and Surgeons and 5Department of Epidemiology, Mailman School of Public Health, Columbia University, New York, New York; 6Department of Cancer Biology, Dana-Farber Cancer Institute; 7Department of Surgery, Brigham and Women’s Hospital; 8Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts; 9Department of Systems Biology, University of Texas M. D. Anderson Cancer Center, Houston, Texas; 10Seina¨joki Central Hospital, Seina¨joki, Finland; 11Institute of Medical Technology, University and University Hospital of Tampere, Tampere, Finland; and 12Pathology Unit, Mediterranean Institute of Oncology, Catania, Italy Abstract Introduction Lesions of ERBB2, PTEN, and PIK3CA activate the phosphati- ERBB2/HER2/Neu (human epidermal growth factor receptor 2), dylinositol 3-kinase (PI3K) pathway during cancer development PTEN (phosphatase and tensin homologue deleted on chromo- by increasing levels of phosphatidylinositol-3,4,5-triphosphate some ten), and PIK3CA (encodes the p110a subunit of phospha- (PIP3). -

Serum and Glucocorticoid-Regulated Kinase 1 (SGK1)
ONCOLOGY REPORTS 39: 1505-1515, 2018 Serum and glucocorticoid-regulated kinase 1 (SGK1) is a predictor of poor prognosis in non-small cell lung cancer, and its dynamic pattern following treatment with SGK1 inhibitor and γ-ray irradiation was elucidated ZHIYUAN TANG1*, QIN SHEN1*, HAO XIE2, ZHU ZHOU3, GUANGLIN SHI1, CAIXIN ZHANG1, ANAZ MOHAMMED1, YI WU1, SONGSHI NI1 and XIAOYU ZHOU1 1Department of Respiratory Medicine, The Affiliated Hospital of Nantong University, Nantong, Jiangsu 226001; 2Key Laboratory of Drug Metabolism and Pharmacokinetics, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing, Jiangsu, 210009, P.R. China; 3Thomas J. Long School of Pharmacy and Health Sciences, University of the Pacific, Stockton, CA 95211, USA Received March 24, 2017; Accepted December 21, 2017 DOI: 10.3892/or.2018.6181 Abstract. The tumor suppressor gene p53 and its dynamic Introduction patterns have caused widespread attention in the field of cancer research. Serum and glucocorticoid-regulated Lung cancer is the leading cause of cancer-related death kinase 1 (SGK1) with features of serine/threonine kinase with an increasingly higher mortality and incidence among activity, which also contributes to the structural and func- younger individuals worldwide and it has become one of the tional similarities with the AKT family of kinases, is a key most serious threats to the quality of human life (1,2). In recent enzyme in the regulation of immune responses in tumor decades, the treatment of lung cancer has made great progress cells, and SGK1 was noted to be expressed in close relation yet the prognosis remains poor with a 5-year survival rate to p53 protein levels, and there exists a negative feedback less than 15%, and a median survival time of 16 to 18 months pathway between intracellular SGK1 and p53. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Serum- and Glucocorticoid-Inducible Kinase 1 and the Response to Cell Stress
Review www.cell-stress.com Serum- and glucocorticoid-inducible kinase 1 and the response to cell stress Florian Lang1,*, Christos Stournaras2, Nefeli Zacharopoulou2, Jakob Voelkl3,4, Ioana Alesutan3,4,5 1 Department of Vegetative and Clinical Physiology, Eberhard-Karls-University, Tübingen, Germany. 2 Department of Biochemistry, University of Crete Medical School, Voutes, Heraklion, Greece- 3 Department of Internal Medicine and Cardiology, Charité – Universitätsmedizin Berlin, Germany. 4 DZHK (German Centre for Cardiovascular Research), partner site Berlin, Germany. 5 Berlin Institute of Health (BIH), Berlin, Germany. * Corresponding Author: Florian Lang, MD, Department of Physiology, University of Tübingen, Wilhelmstr. 56, 72076 Tübingen, Germany; Phone +49-7071 29 72194; Fax: +49-7071 29 5618; E-mail: [email protected] ABSTRACT Expression of the serum- and glucocorticoid-inducible kinase 1 doi: 10.15698/cst2019.01.170 (SGK1) is up-regulated by several types of cell stress, such as ischemia, radia- Received originally: 08.06.2018 tion and hyperosmotic shock. The SGK1 protein is activated by a signaling cas- in revised form: 15.10.2018, Accepted 17.10.2018, cade involving phosphatidylinositide-3-kinase (PI3K), 3-phosphoinositide- Published 02.12.2018. dependent kinase 1 (PDK1) and mammalian target of rapamycin (mTOR). + + + + - SGK1 up-regulates Na /K -ATPase, a variety of carriers including Na -,K -,2Cl - cotransporter (NKCC), NaCl cotransporter (NCC), Na+/H+ exchangers, diverse Keywords: SGK1, glycolysis, cell amino acid transporters and several glucose carriers such as Na+-coupled glu- survival, cell migration, angiogenesis, cose transporter SGLT1. SGK1 further up-regulates a large number of ion fibrosis and tissue calcification. + + channels including epithelial Na channel ENaC, voltage-gated Na channel 2+ 2+ SCN5A, Ca release-activated Ca channel (ORAI1) with its stimulator STIM1, Abbreviations: epithelial Ca2+ channels TRPV5 and TRPV6 and diverse K+ channels. -

SGK1 Activation Exacerbates Diet-Induced Obesity, Metabolic Syndrome and Hypertension
244 1 Journal of C Sierra-Ramos, SGK1 and metabolic syndrome 244:1 149–162 Endocrinology S Velázquez-García et al. development RESEARCH SGK1 activation exacerbates diet-induced obesity, metabolic syndrome and hypertension Catalina Sierra-Ramos1,*, Silvia Velazquez-Garcia1,*, Arianna Vastola-Mascolo1, Guadalberto Hernández1, Nourdine Faresse2,† and Diego Alvarez de la Rosa1 1Department of Basic Medical Sciences, Institute of Biomedical Technologies and Center for Biomedical Research of the Canary Islands (CIBICAN), Universidad de La Laguna, Tenerife, Spain 2Institute of Anatomy, University of Zurich, Zurich, Switzerland Correspondence should be addressed to D Alvarez de la Rosa: [email protected] *(C Sierra-Ramos and S Velázquez-García contributed equally to this work) †(N Faresse is now at DIVA Expertise, 1 Place Pierre Potier, Toulouse, France) Abstract The serum- and glucocorticoid-induced kinase 1 (SGK1) is a transcriptional target Key Words of steroid hormones including glucocorticoids or aldosterone in addition to other f insulin resistance stimuli such as glucose. SGK1 is activated via phosphoinositide 3-kinase, placing it f blood pressure downstream of insulin signaling. SGK1 participates in the upregulation of kidney Na+ f serum and glucocorticoid- reabsorption by aldosterone and has been linked to obesity-related hypertension induced kinase in humans. We hypothesized that a systemic increase in SGK1 activity may trigger a f mouse model multiplicity of mechanisms leading to simultaneous development of the main conditions f fatty liver that characterize the metabolic syndrome (MetS), including hypertension. We used a transgenic mouse model made with a bacterial artificial chromosome containing the whole mouse Sgk1 gene modified to introduce an activating point mutation.