Genomic History of the Sardinian Population
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Allevamento Del Suino, Produzione E Conservazione Dei Salumi
Dipartimento per le produzioni zootecniche Servizio produzioni zootecniche Corso teorico-pratico Allevamento del suino, produzione e conservazione dei salumi Attività di formazione rivolta ai gestori di agriturismi, fattorie didattiche e allevatori della Gallura Programma delle lezioni e calendario degli incontri Sportello Unico Territoriale dell’Alta Ogliastra Via N. Bixio n. 2, Tortolì - Tel. 0782 623084, fax 0782 628033 Data SUT Sede corso Argomento 13-ott-11 Alta Ogliastra Arzana Presentazione Corso Situazione regionale della filiera suinicola Tipologia e sistemi di allevamento dei suini 18-ott-11 Alta Ogliastra Arzana Le razze Gestione dell'allevamento Organizzazione delle varie fasi 20-ott-11 Alta Ogliastra Arzana Gestione sanitaria: Principali malattie Biosicurezza in allevamento 25-ott-11 Alta Ogliastra Arzana Alimentazione: Gli alimenti e le sostanze nutritive Principi di razionamento 27-ott-11 Alta Ogliastra Arzana Lezione pratica Foresta Burgos. Il suino di razza sarda: storia, attualità e prospettive; visita allevamento all'aperto dell'AGRIS. Eventuale visita guidata ad altro/i allevamento/i. 3-nov-11 Alta Ogliastra Arzana Macellazione, qualità delle carni e tagli 8-nov-11 Alta Ogliastra Arzana Classificazione dei salumi. Materie prime per la produzione dei salumi: caratteristiche fisiche, chimiche e microbiologiche; Additivi, aromi e starter 10-nov-11 Alta Ogliastra Arzana Lezione pratica sulla trasformazione e lavorazione dei salumi 15-nov-11 Alta Ogliastra Arzana La macellazione familiare e in agriturismo, La trasformazione artigianale: Iter autorizzativo e caratteristiche dei locali Procedure autorizzative per l'esportazione delle carni suine al di fuori della regione 17-nov-11 Alta Ogliastra Arzana La multifunzionalità dell'azienda agricola 22-nov-11 Alta Ogliastra Arzana Valutazione dei salumi: Analisi sensoriale Principali difetti Sportello Unico Territoriale della Barbagia Via De Gasperi, Gavoi - Tel. -

Elenco Corse
Centro Interuniversitario Regione Autonoma della Sardegna Provincia dell’Ogliastra Ricerche economiche e mobilità PROVINCIA DELL’OGLIASTRA ELENCO CORSE PROGETTO DEFINITIVO Centro Interuniversitario Regione Autonoma della Sardegna Provincia dell’Ogliastra Ricerche economiche e mobilità UNIVERSITÁ DEGLI STUDI DI CAGLIARI CIREM CENTRO INTERUNIVERSITARIO RICERCHE ECONOMICHE E MOBILITÁ Responsabile Scientifico Prof. Ing. Paolo Fadda Coordinatore Tecnico/Operativo Dott. Ing. Gianfranco Fancello Gruppo di lavoro Ing. Diego Corona Ing. Giovanni Durzu Ing. Paolo Zedda Centro Interuniversitario Regione Autonoma della Sardegna Provincia dell’Ogliastra Ricerche economiche e mobilità SCHEMA DEI CORRIDOI ORARIO CORSE CORSE ‐ OGLIASTRA ‐ ANDATA RITORNO CORRIDOIO "GENNA e CRESIA" CORRIDOIO "GENNA e CRESIA" Partenza Arrivo Nome Linee Nome Corsa Km Origine Destinazione Tipologia Veicolo Partenza Arrivo Nome Linee Nome Corsa Km Origine Destinazione Tipologia Veicolo 06:15 07:25 LINEA ROSSA 3630 C1 ‐ fer 40,151 JERZU ARBATAX BUS_15_POSTI A 17:40 18:50 LINEA ROSSA 3630 C1 ‐ fer 40,151 ARBATAX JERZU BUS_15_POSTI R 06:50 08:00 LINEA ROSSA 3630 C ‐ scol 43,012 OSINI NUOVO TORTOLI' STAZIONE FDS BUS_55_POSTI A 13:40 14:50 LINEA ROSSA 3630 C ‐ scol 43,012 TORTOLI' STAZIONE FDS OSINI NUOVO BUS_55_POSTI R 07:00 07:45 LINEA ROSSA 3511 C2 ‐ scol 28,062 LANUSEI PIAZZA V. EMANUELE1 JERZU BUS_55_POSTI A 13:42 14:27 LINEA ROSSA 3511 C2 ‐ scol 28,062 JERZU LANUSEI PIAZZA V. EMANUELE1 BUS_55_POSTI R 07:15 08:00 LINEA ROSSA 3511 C1 ‐ scol 28,062 JERZU LANUSEI PIAZZA V. EMANUELE1 BUS_55_POSTI A 13:42 14:27 LINEA ROSSA 3511 C1 ‐ scol 28,062 LANUSEI PIAZZA V. EMANUELE1 JERZU BUS_55_POSTI R 07:30 07:58 LINEA ROSSA 3 C ‐ scol 18,316 BARI SARDO LANUSEI PIAZZA V. -

N.001-2019 Istituz SUAPE Presso Unione Comuni D'ogliastra
COMUNE DI CARDEDU PROVINCIA DI NUORO DELIBERAZIONE DEL CONSIGLIO COMUNALE n. 1 del 24/01/2019 COPIA Oggetto: Istituzione dello Sportello Unico per le Attività Produttive e per l'Attività Edilizia SUAPE Associato Ogliastra 2, presso lUnione Comuni d'Ogliastra. Approvazione schema di convenzione (allegato A). Approvazione schema di regolamento (allegato B). L’anno DUEMILADICIANNOVE il giorno ventiquattro del mese di gennaio alle ore 18,05 presso la sala delle adunanze, si è riunito il Consiglio Comunale,convocato con avvisi spediti a termini di legge, in sessione ordinaria ed in prima convocazione. Risultano presenti/assenti i seguenti consiglieri: PIRAS MATTEO PRESENTE MOLINARO ARMANDO PRESENTE COCCO SABRINA PRESENTE PILIA PATRIK PRESENTE CUCCA PIER LUIGI PRESENTE PISU MARIA SOFIA ASSENTE CUCCA SIMONE PRESENTE PODDA MARCO PRESENTE DEMURTAS MARCO PRESENTE SCATTU FEDERICO PRESENTE LOTTO GIOVANNI PRESENTE VACCA MARCELLO PRESENTE MARCEDDU MIRCO ASSENTE Quindi n. 11 (undici) presenti su n. 13 (tredici) componenti assegnati, n. 2 (due) assenti. il Signor Matteo Piras, nella sua qualità di Sindaco, assume la presidenza e, assistito dal Segretario Comunale Dott.ssa Giovannina Busia, sottopone all'esame del Consiglio la proposta di deliberazione di cui all'oggetto, di seguito riportata: IL CONSIGLIO COMUNALE PREMESSO che: • i Comuni di Arzana, Bari Sardo, Cardedu, Elini, Ilbono, Lanusei e Loceri, con rispettive deliberazioni consiliari, si sono costituiti in Unione ai sensi dell’art. 32 del T.U.E.L. 267/2000 e dell’articolo 3 della ex Legge Regionale 12/2005 - oggi sostituita dalla Legge Regionale 2/2016 - denominandola “ Unione Comuni D’Ogliastra ” ed approvando con i medesimi atti lo Statuto e l’atto costitutivo dell’Unione; • con deliberazione del Consiglio comunale n. -

Autorizzazione Societa GLE Sport A.S.D. Per
PROVINCIA DI NUORO SETTORE INFRASTRUTTURE ZONA OMOGENEA OGLIASTRA SERVIZIO AGRICOLTURA, MANUTENZIONI E TUTELA DEL TERRITORIO DETERMINAZIONE N° 617 DEL 14/06/2019 OGGETTO: Autorizzazione societa GLE Sport A.S.D. per lo svolgimento di una competizione amatoriale e dilettantistica denominata " 8° Rally di Sardegna Bike " indetta per i giorni 16 _ 17 _ 18 _ 19 _ 20 _ 21 Giugno 2019 _ internazionale di Rally _ Raid di Mountain Bike a Tappe. IL RESPONSABILE DEL SERVIZIO VISTI: - l’art.65, comma 2, lett.B) della L.R. 9/2007 di trasferimento alla Provincia delle competenze relative al rilascio delle autorizzazioni in materia di competizioni sportive su strada; - la nota prot. 275 del 30 gennaio 2008 dell’Assessorato Regionale dei Lavori Pubblici, in base alla quale i provvedimenti autorizzativi per lo svolgimento di manifestazioni sportive su strada sono demandati alle Amministrazioni Provinciali competenti per territorio ai sensi della citata L.R. N.9/2006; - l’istanza assunta al protocollo della Provincia di Nuoro Zona Omogenea dell’Ogliastra al n. 2217 del 15/05/2019 della Società Sportiva G.L.E. Sport A.S.D., con sede in Cagliari in via Farina n° 11, a firma del suo Presidente, Sig. Gian Domenico Nieddu, per ottenere l’autorizzazione allo svolgimento di una manifestazione/gara amatoriale/dilettantistica denominata “8° Rally di Sardegna Bike” – internazionale di Rally-Raid di Mountain Bike a tappe da tenersi nei territori dei cantieri dell’Ente Forestas ricadenti nei Comuni di Arzana, Baunei, Desulo, Gairo, Orgosolo, Seui, Talana, Triei, Urzulei, Ussassai e Villagrande Strisaili nei giorni 16 – 17 – 18 – 19 – 20 – 21 Giugno 2019 . -

Official Journal C 3 of the European Union
Official Journal C 3 of the European Union Volume 62 English edition Information and Notices 7 January 2019 Contents IV Notices NOTICES FROM EUROPEAN UNION INSTITUTIONS, BODIES, OFFICES AND AGENCIES European Commission 2019/C 3/01 Euro exchange rates .............................................................................................................. 1 NOTICES FROM MEMBER STATES 2019/C 3/02 Notice inviting proposals for the transfer of an extraction area for hydrocarbon extraction ................ 2 2019/C 3/03 Communication from the Minister for Economic Affairs and Climate Policy of the Kingdom of the Netherlands pursuant to Article 3(2) of Directive 94/22/EC of the European Parliament and of the Council on the conditions for granting and using authorisations for the prospection, exploration and production of hydrocarbons ................................................................................................... 4 V Announcements PROCEDURES RELATING TO THE IMPLEMENTATION OF COMPETITION POLICY European Commission 2019/C 3/04 Prior notification of a concentration (Case M.9148 — Univar/Nexeo) — Candidate case for simplified procedure (1) ........................................................................................................................ 6 EN (1) Text with EEA relevance. OTHER ACTS European Commission 2019/C 3/05 Publication of the amended single document following the approval of a minor amendment pursuant to the second subparagraph of Article 53(2) of Regulation (EU) No 1151/2012 ............................. -

Presentazione Primavera Nel Marghine, Ogliastra E Baronia 2016
Primavera nel Marghine, Ogliastra e Baronia Edizione 2016 Comunicato stampa Nuoro, 13 aprile. Dal 16 aprile sino al 26 giugno 2016 si rinnova il consueto appuntamento primaverile che dal 2006 propone il viaggio tra i borghi e i paesi del Cuore della Sardegna. Alla conferenza stampa di presentazione della decima edizione della Primavera nel Marghine, Ogliastra e Baronia hanno partecipato il Presidente della CCIAA di Nuoro Agostino Cicalò, dell’Aspen Vincenzo Cannas e i rappresentanti di alcuni dei comuni aderenti al circuito (Lotzorai, Lodè, Posada, Urzulei, Bortigali, Bari Sardo, Arbatax – Tortolì, Girasole, Loceri). La Primavera è un lungo percorso che vedrà protagonisti venti paesi dalle affascinanti bellezze naturali e paesaggistiche, antiche tradizioni e un ricco patrimonio enogastronomico. Nel calendario della Primavera si confermano alcuni paesi dello scorso anno, come Girasole, Lodè, Siniscola, Bosa, Triei, Lotzorai, Macomer, Baunei, Arbatax, Bari Sardo, Urzulei e Lanusei che metteranno in luce le particolarità e le eccellenze dei diversi territori. Tra le novità dell’edizione 2016 si segnala la partecipazione di Loceri, Ilbono, Sindia, Posada, Lei, Osini, Bortigali e Birori. Il Viaggio nel Cuore della Sardegna in Primavera propone soprattutto la scoperta dei paesaggi incantati e della natura incontaminata, tra i colori intensi della natura e la costa dalle numerose varietà del turchese. “Fino a quando l’Aspen e la Camera di Commercio potranno proseguire nelle proprie attività - dichiara il Presidente Cannas - il nostro impegno sarà quello di continuare ad investire nelle manifestazioni utili per allungare la stagione turistica. Tutto ciò è possibile grazie al coordinamento con le amministrazioni comunali e con le realtà locali che vantano una particolare bravura nella realizzazione di prodotti di artigianato e dell’enogastronomia”. -

Filoloxía Asturiana
REVISTA DE Filoloxía Asturiana volume 11/12 - años 2011/2012 Revista de Filoloxía Asturiana Revista de Filoloxía Asturiana (Anuariu universitariu d’estudios llingüísticos y lliterarios asturianos y románicos) Edita: Grupu d’Investigación Seminariu de Filoloxía Asturiana Universidá d’Uviéu Director: Xulio Viejo Fernández Secretaria: Taresa Fernández Lorences Comité de Redacción Fernando Álvarez-Balbuena García (Dptu. Filoloxía Clásica y Románica, Universidá d’Uviéu), Ramón d’Andrés Díaz (Dptu. Filoloxía Española, Universidá d’Uviéu), Xuan Carlos Busto Cortina (Dptu. Filoloxía Clásica y Románica, Universidá d’Uviéu), María Cueto Fernández (Dptu. Filoloxía Española, Universidá d’Uviéu), Iván Cuevas, Taresa Fernández Lorences (Dptu. Filoloxía Española, Universidá d’Uviéu), Roberto Hinojal Díaz (Dptu. Filoloxía Española, Universidá d’Uviéu), Rosa María Medina Granda (Dptu. Filoloxía Clásica y Románica, Universidá d’Uviéu), Leopoldo Sánchez Torre (Dptu. Filoloxía Española, Universidá d’Uviéu), Xulio Viejo Fernández (Dptu. Filoloxía Española, Universidá d’Uviéu) Miembros del Seminariu de Filoloxía Asturiana de la Universidá d’Uviéu Comité Científicu Rosario Álvarez (Universidade de Santiago de Compostela-Instituto da Lingua Galega), Antonio Bárbolo Alves (CEL-Universidade de Trás-os-Mon- tes e Alto Douro, Portugal), Eduardo Blasco Ferrer (Università di Cagliari, Cerdeña), Inés Fernández Ordóñez (Universidad Autónoma de Madrid-Real Academia Española), José Enrique Gargallo Gil (Universitat de Barcelona), Hans Goebl (Universität Salzburg, Austria), -

Statistical Bulletin 20 17 1 - Quarter Quarter 1
quarter 1 - 2017 Statistical Bulletin quarter 1 Statistical Bulletin Statistical publications and distribution options Statistical publications and distribution options The Bank of Italy publishes a quarterly statistical bulletin and a series of reports (most of which are monthly). The statistical information is available on the Bank’s website (www.bancaditalia.it, in the Statistical section) in pdf format and in the BDS on-line. The pdf version of the Bulletin is static in the sense that it contains the information available at the time of publication; by contrast the on-line edition is dynamic in the sense that with each update the published data are revised on the basis of any amendments received in the meantime. On the Internet the information is available in both Italian and English. Further details can be found on the Internet in the Statistics section referred to above. Requests for clarifications concerning data contained in this publication can be sent by e-mail to [email protected]. The source must be cited in any use or dissemination of the information contained in the publications. The Bank of Italy is not responsible for any errors of interpretation of mistaken conclusions drawn on the basis of the information published. Director: GRAZIA MARCHESE For the electronic version: registration with the Court of Rome No. 23, 25 January 2008 ISSN 2281-4671 (on line) Notice to readers Notice to readers I.The appendix contains methodological notes with general information on the statistical data and the sources form which they are drawn. More specific notes regarding individual tables are given at the foot of the tables themselves. -

O D Infanzia 2013-14 Da Pubblicare Per Decreto Organico
UFFICIO SCOLASTICO PROVINCIALE NUORO DETERMINAZIONE ORGANICO DI DIRITTO ANNO SCOLASTICO 2013/2014 ISTITUTO COMUNE SEDE DELLA SCUOLA SEZ. POSTI POSTI TOTALE PRINCIPALE SEZIONE SOSTEGNO ORG.FUNZ. 1 ATZARA ATZARA Via Lamarmora 12 10 ATZARA AUSTIS Via Roma 1 2 ATZARA MEANA SARDO Corso G.M. Agus 2 4 ATZARA SORGONO Via F.lli Costa 1 2 Totali 5 10 2 BARISARDO BARISARDO Via Mannironi 5 10 1 14 BARISARDO LOCERI Via Nazario Sauro 2 4 Totali 7 14 3 BAUNEI BAUNEI Via Lombardo Radice 2 4 1 18 BAUNEI TRIEI Viale Europa, 4 2 4 BAUNEI BAUNEI S.MARIA S. Maria Navarrese 3 6 BAUNEI URZULEI Via Grazia Deledda 2 4 Totali 9 18 4 BITTI BITTI Via San Tomaso 2 4 1 10 BITTI LULA Via A. Gramsci 2 4 BITTI ONANI' Via Cavour 1 2 BITTI ORUNE Via Cuccuretteti 3 6 Totali 8 16 5 BOLOTANA BOLOTANA Via Della Libertà 2 4 0 10 BOLOTANA SILANUS Via Stazione 2 4 BOLOTANA LEI Piazza Municipio 1 2 Totali 5 10 6 BUDONI BUDONI Via Nazionale 5 10 1 10 Totali 5 10 UFFICIO SCOLASTICO PROVINCIALE NUORO DETERMINAZIONE ORGANICO DI DIRITTO ANNO SCOLASTICO 2013/2014 ISTITUTO COMUNE SEDE DELLA SCUOLA SEZ. POSTI POSTI TOTALE PRINCIPALE SEZIONE SOSTEGNO ORG.FUNZ. 7 DESULO DESULO Ovolaccio - V.Cagliari 2 4 1 12 DESULO DESULO Issiria - Via A. Gramsci 2 4 DESULO ARITZO Via Nuova 1 2 DESULO GADONI Via Cui Polla Raffaela 1 2 Totali 6 12 8 DORGALI DORGALI Via Enrico Fermi 2 4 1 20 DORGALI DORGALI Via Lamarmora 2 4 DORGALI DORGALI Via Fratelli Cervi 3 6 DORGALI DORGALI Cala Gonone - Via Grotte 3 6 Totali 10 20 9 FONNI FONNI Via Papa Giovanni 3 6 1 6 Totali 3 6 10 GAIRO GAIRO Via Sardegna 2 4 1 10 GAIRO CARDEDU Via Amsicora 3 6 Totali 5 10 11 GAVOI GAVOI Via Cagliari 36 10 GAVOI OLLOLAI Via Repubblica 2 4 Totali 5 10 12 ILBONO ILBONO Via Dettori 3 6 2 14 ILBONO ELINI Via Grazia Deledda 1 2 ILBONO ARZANA Piazza Roma 3 6 Totali 7 14 13 IRGOLI IRGOLI Via Roma 3 6 0 6 IRGOLI GALTELLI' Via Nazionale 3 6 6 UFFICIO SCOLASTICO PROVINCIALE NUORO DETERMINAZIONE ORGANICO DI DIRITTO ANNO SCOLASTICO 2013/2014 ISTITUTO COMUNE SEDE DELLA SCUOLA SEZ. -

APPALTI AGGIUDICATI (Art
Asmel Consortile S.C. a r.l. Sede Legale: Piazza del Colosseo, 4 – Roma Sede Operativa: Centro Direzionale - Isola G1 - Napoli P.Iva: 12236141003 www.asmecomm.it – portale di committenza pubblica COMUNE DI OVODDA (NU) COMUNE DI OVODDA PROVINCIA DI NUORO AREA TECNICO, MANUTENTIVA e di VIGILANZA | PI: 00914600911 - CF: 80003610914 | Via Sassari n° 4, 08020 Ovodda (NU) | 0784.54023 0784.54475 www.comune.ovodda.nu.it [email protected] PEC: [email protected] Prot. n. 2000 del 10.04.2017 Albo pretorio n. 172 del 10.04.2017 BANDO DI GARA MEDIANTE PROCEDURA APERTA Esecuzione di lavori sulla base del progetto esecutivo. Art. 26, comma 8, D.Lgs n. 50 del 2016 PIA NU 13-14: “Completamento piano degli insediamenti produttivi PIP Ghiliddoe - opere di viabilità” CUP: E84H12000020002 CIG: 6888258253 AVVISO RELATIVO AGLI APPALTI AGGIUDICATI (Art. 98 D.Lgs 50/2016 - Allegato XIV, Parte I, lettera D) 1. Nome, numero di identificazione, ove previsto, indirizzo comprensivo di codice NUTS, telefono, fax, posta elettronica e indirizzo Internet dell’amministrazione aggiudicatrice e, se diverso, del servizio al quale rivolgersi per informazioni complementari. = Comune di Ovodda - Via Sassari n. 4 - 08020 Ovodda (NU) Tel. 0784/54023 – Fax 0784/54475 – info@ comune.ovodda.nu.it – PEC: [email protected] - www.comune.ovodda.nu.it 2. Tipo di amministrazione aggiudicatrice e principale attività esercitata. = Ente Locale. 3. Se del caso, l’indicazione che l’amministrazione aggiudicatrice è una centrale di committenza o che si tratta di una qualsiasi altra forma di appalto congiunto. = Asmel Consortile S.C. a r.l. -
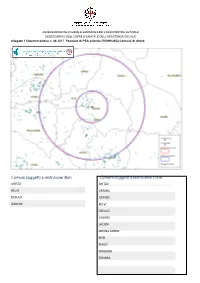
Comuni Soggetti a Restrizione 3Km Comuni Soggetti a Restrizione 10Km
ASSESSORADU DE S’IGIENE E SANIDADE E DE S’ASSISTÈNTZIA SOTZIALE ASSESSORATO DELL’IGIENE E SANITA’ E DELL’ASSISTENZA SOCIALE Allegato 1 Determinazione n. 88.2017 Focolaio di PSA azienda IT001NU054 Comune di Aritzo Comuni soggetti a restrizione 3km Comuni soggetti a restrizione 10km ARITZO ARITZO BELVI ARZANA DESULO ATZARA GADONI BELVI DESULO GADONI LACONI MEANA SARDO SEUI SEULO SORGONO TONARA Alleva ment i in zon a di res tri zione 3KM Asl Comune Codice Denominazione Z103 ARITZO 001NU001 CASULA SALVATORE NU001AA Z103 ARITZO 001NU009 MANCA BONARIA Z103 ARITZO 001NU016 MELIS TEODORA Z103 ARITZO 001NU023 MANCA FRANCESCO Z103 ARITZO 001NU024 FRAU DAVIDE ANTIOCO Z103 ARITZO 001NU035 PABA ANTONIO 001BQ Z103 ARITZO 001NU039 LOI PIERO 001BV Z103 ARITZO 001NU046 MANCA ANTONIO Z103 ARITZO 001NU050 ORRU' SALVATORE Z103 ARITZO 001NU051 PORCEDDA ANTONELLO Z103 ARITZO 001NU053 PRANTEDDU ARMANDO Z103 ARITZO 001NU054 MUGGIRONI ALESSANDRO Z103 ARITZO 001NU056 PABA MARCO Z103 ARITZO 001NU058 PABA SEBASTIANO Z103 ARITZO 001NU062 FRAU SALVATORE Z103 ARITZO 001NU063 MAXIA MASSIMILIANO Z103 ARITZO 001NU064 PILI SERENA Z103 ARITZO 001NU070 GARAU GIAMPAOLO Z103 ARITZO 001NU071 SECCI FRANCO Z103 ARITZO 001NU072 ONANO EMANUELE Z103 ARITZO 001NU075 LEONE ANTONIO Z103 ARITZO 001NU076 FRAU IGNAZIO Z103 ARITZO 001NU077 PRANTEDDU FRANCESCO 16.02.1956 Z103 ARITZO 001NU078 FIGUS ANTONIO ANTIOCO Z103 ARITZO 001NU084 PABA MICHELANGELO Z103 ARITZO 001NU085 ATZORI NATALINA Z103 ARITZO 001NU086 PILI FRANCESCO Z103 ARITZO 001NU086 PILI GIOVANNI Z103 ARITZO 001NU087 LOI ANTONIO -

Provincia Di Nuoro Amministratore Straordinario
PROVINCIA DI NUORO AMMINISTRATORE STRAORDINARIO L.R. N. 02 DEL 04 FEBBRAIO 2016 __________ DELIBERAZIONE N. 69 del 03/05/2019 OGGETTO: Accordo di collaborazione per la attivazione di un servizio di mediazione culturale nei centri di accoglienza del CPIA n. 3 di Nuoro. L'anno 2019, addì 03, del mese di Maggio, alle ore 13:20 presso la sede provinciale, Piazza Italia, 22 è presente il Sig. Costantino Tidu nominato Amministratore Straordinario della Provincia di Nuoro. Partecipa il Vice Segretario Provinciale Dott. Giuseppe Zucca. Provincia di Nuoro – Deliberazione n. 69 del 03/05/2019 1 L’AMMINISTRATORE STRAORDINARIO VISTA l’allegata proposta di deliberazione concernente l’oggetto; RICHIAMATA la Legge Regionale Sardegna n° 2 del 04/02/2016 di riordino del sistema delle Autonomie locali della Sardegna con la quale è stata prevista la nomina dell’Amministratore Straordinario della Provincia di Nuoro, come modificata con L.R. n. 5 del 13.04.2017; VISTA la deliberazione della Giunta Regionale n. 69/12 del 23.12.2016, con la quale il Sig. Costantino Tidu è stato nominato Amministratore Straordinario della Provincia di Nuoro, con funzioni estese anche alla cessata Provincia Ogliastra; VISTO il vigente Statuto della Provincia di Nuoro; VISTO l’art. 24, comma 7 della L.R. n. 2 del 04/02/2016 che definisce le funzioni dell'Amministratore Straordinario; VISTI i seguenti pareri espressi ai sensi del comma 1 dell’art.49 del T.U. 18/08/2000 n. 267 sulla proposta in argomento: - dal Dirigente del Settore Affari Istituzionali e Programmazione: “Favorevole