Full-Text PDF (Final Published Version)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evolution of the Patellar Sesamoid Bone in Mammals
A peer-reviewed version of this preprint was published in PeerJ on 21 March 2017. View the peer-reviewed version (peerj.com/articles/3103), which is the preferred citable publication unless you specifically need to cite this preprint. Samuels ME, Regnault S, Hutchinson JR. 2017. Evolution of the patellar sesamoid bone in mammals. PeerJ 5:e3103 https://doi.org/10.7717/peerj.3103 Evolution of the patellar sesamoid bone in mammals Mark E Samuels 1, 2 , Sophie Regnault 3 , John R Hutchinson Corresp. 3 1 Department of Medicine, University of Montreal, Montreal, Quebec, Canada 2 Centre de Recherche du CHU Ste-Justine, Montreal, Quebec, Canada 3 Structure & Motion Laboratory, Department of Comparative Biomedical Sciences, The Royal Veterinary College, Hatfield, Hertfordshire, United Kingdom Corresponding Author: John R Hutchinson Email address: [email protected] The patella is a sesamoid bone located in the major extensor tendon of the knee joint, in the hindlimb of many tetrapods. Although numerous aspects of knee morphology are ancient and conserved among most tetrapods, the evolutionary occurrence of an ossified patella is highly variable. Among extant (crown clade) groups it is found in most birds, most lizards, the monotreme mammals and almost all placental mammals, but it is absent in most marsupial mammals as well as many reptiles. Here we integrate data from the literature and first-hand studies of fossil and recent skeletal remains to reconstruct the evolution of the mammalian patella. We infer that bony patellae most likely evolved between four to six times in crown group Mammalia: in monotremes, in the extinct multituberculates, in one or more stem-mammal genera outside of therian or eutherian mammals, and up to three times in therian mammals. -

Digital Reconstruction of the Inner Ear of Leptictidium Auderiense
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by RERO DOC Digital Library Published in "Paläontologische Zeitschrift 90(1): 153–171, 2016" which should be cited to refer to this work. Digital reconstruction of the inner ear of Leptictidium auderiense (Leptictida, Mammalia) and North American leptictids reveals new insight into leptictidan locomotor agility Irina Ruf1,2 • Virginie Volpato1,3 • Kenneth D. Rose4 • Guillaume Billet2,5 • Christian de Muizon5 • Thomas Lehmann1 Abstract Leptictida are basal Paleocene to Oligocene semicircular canals than the leptictids under study. Our eutherians from Europe and North America comprising estimations reveal that Leptictidium was a very agile ani- species with highly specialized postcranial features mal with agility score values (4.6 and 5.5, respectively) including elongated hind limbs. Among them, the Euro- comparable to Macroscelidea and extant bipedal saltatory pean Leptictidium was probably a bipedal runner or jum- placentals. Leptictis and Palaeictops have lower agility per. Because the semicircular canals of the inner ear are scores (3.4 to 4.1), which correspond to the more gener- involved in detecting angular acceleration of the head, their alized types of locomotion (e.g., terrestrial, cursorial) of morphometry can be used as a proxy to elucidate the agility most extant mammals. In contrast, the angular velocity in fossil mammals. Here we provide the first insight into magnitude predicted from semicircular canal angles sup- inner ear anatomy and morphometry of Leptictida based on ports a conflicting pattern of agility among leptictidans, but high-resolution computed tomography of a new specimen the significance of these differences might be challenged of Leptictidium auderiense from the middle Eocene Messel when more is known about intraspecific variation and the Pit (Germany) and specimens of the North American pattern of semicircular canal angles in non-primate Leptictis and Palaeictops. -

The Evolution of Micro-Cursoriality in Mammals
© 2014. Published by The Company of Biologists Ltd | The Journal of Experimental Biology (2014) 217, 1316-1325 doi:10.1242/jeb.095737 RESEARCH ARTICLE The evolution of micro-cursoriality in mammals Barry G. Lovegrove* and Metobor O. Mowoe* ABSTRACT Perissodactyla) in response to the emergence of open landscapes and In this study we report on the evolution of micro-cursoriality, a unique grasslands following the Eocene Thermal Maximum (Janis, 1993; case of cursoriality in mammals smaller than 1 kg. We obtained new Janis and Wilhelm, 1993; Yuanqing et al., 2007; Jardine et al., 2012; running speed and limb morphology data for two species of elephant- Lovegrove, 2012b; Lovegrove and Mowoe, 2013). shrews (Elephantulus spp., Macroscelidae) from Namaqualand, Loosely defined, cursorial mammals are those that run fast. South Africa, which we compared with published data for other However, more explicit definitions of cursoriality remain obscure mammals. Elephantulus maximum running speeds were higher than because locomotor performance is influenced by multiple variables, those of most mammals smaller than 1 kg. Elephantulus also including behaviour, biomechanics, physiology and morphology possess exceptionally high metatarsal:femur ratios (1.07) that are (Taylor et al., 1970; Garland, 1983a; Garland, 1983b; Garland and typically associated with fast unguligrade cursors. Cursoriality evolved Janis, 1993; Stein and Casinos, 1997; Carrano, 1999). In an in the Artiodactyla, Perissodactyla and Carnivora coincident with evaluation of these definition problems, Carrano (Carrano, 1999) global cooling and the replacement of forests with open landscapes argued that ‘…morphology should remain the fundamental basis for in the Oligocene and Miocene. The majority of mammal species, making distinctions between locomotor performance…’. -

New Large Leptictid Insectivore from the Late Paleogene of South Dakota, USA
New large leptictid insectivore from the Late Paleogene of South Dakota, USA TJ MEEHAN and LARRY D. MARTIN Meehan, T.J. and Martin, L.D. 2012. New large leptictid insectivore from the Late Paleogene of South Dakota, USA. Acta Palaeontologica Polonica 57 (3): 509–518. From a skull and mandible, we describe a new genus and species of a primitive insectivore (Mammalia: Insectivora: Leptictida: Leptictidae). Its large body size and higher−crowned teeth indicate a different feeding ecology from other leptictid insectivores. With evidence of some heavy, flat wear on the molariform teeth, its shift in diet was likely to greater herbivory. Unlike the narrow snout of Blacktops, this new leptictid retains a broad snout, suggesting that small verte− brates were still important dietary components. The specimen was collected from the floodplain deposits of the lower or middle White River Group of South Dakota, which represent the latest Eocene to earliest Oligocene (Chadronian and Orellan North American Land Mammal “Ages”). Key words: Mammalia, Leptictidae, Leptictis, Megaleptictis, Eocene, Oligocene, White River Group, South Dakota, North America. TJ Meehan [[email protected]], Research Associate, Section of Vertebrate Paleontology, Carnegie Museum of Natural History, 4400 Forbes Avenue, Pittsburgh, PA 15213, USA; Larry D. Martin [[email protected]], Division of Vertebrate Paleontology, Natural History Museum and Biodiversity Re− search Center, University of Kansas, Lawrence, KS 66045, USA. Received 4 April 2011, accepted 25 July 2011, available online 17 August 2011. Introduction molariform teeth. A fossa in this region at least suggests in− creased snout mobility, but no definitive anatomical argument Leptictida is a primitive order of placental, insectivorous has been made to support a highly mobile cartilaginous snout mammals convergent to extant sengis or elephant “shrews” tip, as in sengis. -

Afrotherian Conservation – Number 16
AFROTHERIAN CONSERVATION Newsletter of the IUCN/SSC Afrotheria Specialist Group Number 16 Edited by PJ Stephenson September 2020 Afrotherian Conservation is published annually by the measure the effectiveness of SSC’s actions on biodiversity IUCN Species Survival Commission Afrotheria Specialist conservation, identification of major new initiatives Group to promote the exchange of news and information needed to address critical conservation issues, on the conservation of, and applied research into, consultations on developing policies, guidelines and aardvarks, golden moles, hyraxes, otter shrews, sengis and standards, and increasing visibility and public awareness of tenrecs. the work of SSC, its network and key partners. Remarkably, 2020 marks the end of the current IUCN Published by IUCN, Gland, Switzerland. quadrennium, which means we will be dissolving the © 2020 International Union for Conservation of Nature membership once again in early 2021, then reassembling it and Natural Resources based on feedback from our members. I will be in touch ISSN: 1664-6754 with all members at the relevant time to find out who wishes to remain a member and whether there are any Find out more about the Group people you feel should be added to our group. No one is on our website at http://afrotheria.net/ASG.html automatically re-admitted, however, so you will all need to and on Twitter @Tweeting_Tenrec actively inform me of your wishes. We will very likely need to reassess the conservation status of all our species during the next quadrennium, so get ready for another round of Red Listing starting Message from the Chair sometime in the not too distant future. -

Resolving the Relationships of Paleocene Placental Mammals
Biol. Rev. (2015), pp. 000–000. 1 doi: 10.1111/brv.12242 Resolving the relationships of Paleocene placental mammals Thomas J. D. Halliday1,2,∗, Paul Upchurch1 and Anjali Goswami1,2 1Department of Earth Sciences, University College London, Gower Street, London WC1E 6BT, U.K. 2Department of Genetics, Evolution and Environment, University College London, Gower Street, London WC1E 6BT, U.K. ABSTRACT The ‘Age of Mammals’ began in the Paleocene epoch, the 10 million year interval immediately following the Cretaceous–Palaeogene mass extinction. The apparently rapid shift in mammalian ecomorphs from small, largely insectivorous forms to many small-to-large-bodied, diverse taxa has driven a hypothesis that the end-Cretaceous heralded an adaptive radiation in placental mammal evolution. However, the affinities of most Paleocene mammals have remained unresolved, despite significant advances in understanding the relationships of the extant orders, hindering efforts to reconstruct robustly the origin and early evolution of placental mammals. Here we present the largest cladistic analysis of Paleocene placentals to date, from a data matrix including 177 taxa (130 of which are Palaeogene) and 680 morphological characters. We improve the resolution of the relationships of several enigmatic Paleocene clades, including families of ‘condylarths’. Protungulatum is resolved as a stem eutherian, meaning that no crown-placental mammal unambiguously pre-dates the Cretaceous–Palaeogene boundary. Our results support an Atlantogenata–Boreoeutheria split at the root of crown Placentalia, the presence of phenacodontids as closest relatives of Perissodactyla, the validity of Euungulata, and the placement of Arctocyonidae close to Carnivora. Periptychidae and Pantodonta are resolved as sister taxa, Leptictida and Cimolestidae are found to be stem eutherians, and Hyopsodontidae is highly polyphyletic. -

Eutheria (Placental Mammals)
Eutheria (Placental Introductory article Mammals) Article Contents . Introduction J David Archibald, San Diego State University, San Diego, California, USA . Basic Design . Taxonomic and Ecological Diversity Eutheria includes one of three major clades of mammals, the extant members of which are . Fossil History and Distribution referred to as placentals. Phylogeny Introduction have supernumerary teeth (e.g. some whales, armadillos, Eutheria (or Placentalia) is the most taxonomically diverse etc.), in extant placentals the number of teeth is at most of three branches or clades of mammals, the other two three upper and lower incisors, one upper and lower being Metatheria (or Marsupialia) and Prototheria (or canine, four upper and lower premolars, and three upper Monotremata). When named by Gill in 1872, Eutheria and lower molars. Except for one fewer upper molar, a included both marsupials and placentals. It was Huxley in domestic dog retains this pattern. Compared to reptiles, 1880 that recognized Eutheria basically as used today to mammals have fewer skull bones through fusion and loss, include only placentals. McKenna and Bell in their although bones are variously emphasized in each of the Classification of Mammals, published in 1997, chose to three major mammalian taxa. use Placentalia rather than Eutheria to avoid the confusion Physiologically, mammals are all endotherms of varying of what taxa should be included in Eutheria. Others such as degrees of efficiency. They are also homeothermic with a Rougier have used Eutheria and Placentalia in the sense relatively high resting temperature. These characteristics used here. Placentalia includes all extant placentals and are also found in birds, but because of anatomical their most recent common ancestor. -

Review Article Why Is There Discordant Diversity in Sengi
Review article Why is there discordant diversity in sengi (Mammalia: Afrotheria: Macroscelidea) taxonomy and ecology? Galen B. Rathbun* Department of Ornithology and Mammalogy, California Academy of Sciences (San Francisco), c ⁄ o P.O. Box 202, Cambria, CA 93428, U.S.A. allant de de´serts coˆtiers a` des foreˆts de montagne. En raison Abstract de leur isolement sur le continent africain tre`s vite apre`sla The seventeen species of sengis or elephant-shrews form a scission du Gondwana, les sengis ont the´oriquement well-defined clade of mammals endemic to Africa that e´volue´ au de´part sans qu’il existe beaucoup de compe´tition occupy the extremes of terrestrial habitats, from coastal avec les radiations d’autres placentaires. Les caracte´ris- deserts to montane forests. Because of their isolation on tiques de leur histoire incluent de la myrme´cophagie, des Africa soon after the break-up of Gondwanaland, theo- de´placements par bonds, un usage de nids limite´, voire retically sengis initially evolved with little competition inexistant, la monogamie sociale, de petites porte´es de from other placental radiations. Their life history features jeunes pre´coces et l’inexistence de soins maternels pour les include myrmecophagy, saltatorial gaits, no or limited nouveau-ne´s. Toutes ces caracte´ristiques sont uniques use of nests, social monogamy, small litters of precocial pour les Macrosce´lide´s et repre´sentent un regroupement de young and absentee maternal care of neonates. These caracte´ristiques d’habitude associe´es a` de petites antilopes traits together are unique to the Macroscelidea and rep- ou a` des fourmiliers. -

Genomic Evidence Reveals a Radiation of Placental Mammals Uninterrupted
Genomic evidence reveals a radiation of placental PNAS PLUS mammals uninterrupted by the KPg boundary Liang Liua,b,1, Jin Zhangc,1, Frank E. Rheindtd,1, Fumin Leie, Yanhua Que, Yu Wangf, Yu Zhangf, Corwin Sullivang, Wenhui Nieh, Jinhuan Wangh, Fengtang Yangi, Jinping Chenj, Scott V. Edwardsa,k,2, Jin Mengl, and Shaoyuan Wua,m,2 aJiangsu Key Laboratory of Phylogenomics and Comparative Genomics, School of Life Sciences, Jiangsu Normal University, Xuzhou 221116, Jiangsu, China; bDepartment of Statistics & Institute of Bioinformatics, University of Georgia, Athens, GA 30606; cKey Laboratory of High Performance Computing and Stochastic Information Processing of the Ministry of Education of China, Department of Computer Science, College of Mathematics and Computer Science, Hunan Normal University, Changsha, Hunan 410081, China; dDepartment of Biological Sciences, National University of Singapore, Singapore 117543; eKey Laboratory of Zoological Systematics and Evolution, Institute of Zoology, Chinese Academy of Sciences, Beijing 100101, China; fState Key Laboratory of Reproductive Biology & Institute of Zoology, Chinese Academy of Sciences, Beijing 100101, China; gKey Laboratory of Vertebrate Evolution and Human Origins of the Chinese Academy of Sciences, Institute of Vertebrate Paleontology and Paleoanthropology, Chinese Academy of Sciences, Beijing 100044, China; hState Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650223, China; iWellcome Trust Sanger Institute, -
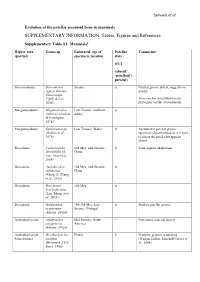
SUPPLEMENTARY INFORMATION: Tables, Figures and References
Samuels et al. Evolution of the patellar sesamoid bone in mammals SUPPLEMENTARY INFORMATION: Tables, Figures and References Supplementary Table S1: Mammals$ Higher taxa Genus sp. Estimated. age of Patellar Comments# (partial) specimen, location state 0/1/2 (absent/ ‘patelloid’/ present) Sinoconodonta Sinoconodon Jurassic 0 Patellar groove absent, suggests no rigneyi (Kielan- patella Jaworowska, Cifelli & Luo, Sinoconodon is included on our 2004) phylogeny within tritylodontids. Morganucodonta Megazostrodon Late Triassic, southern 0 rudnerae (Jenkins Africa & Parrington, 1976) Morganucodonta Eozostrodon sp. Late Triassic, Wales 0 Asymmetric patellar groove, (Jenkins et al., specimens disarticulated so it is hard 1976) to assess the patella but appears absent Docodonta Castorocauda 164 Mya, mid-Jurassic, 0 Semi-aquatic adaptations lutrasimilis (Ji, China Luo, Yuan et al., 2006) Docodonta Agilodocodon 164 Mya, mid-Jurassic, 0 scansorius China (Meng, Ji, Zhang et al., 2015) Docodonta Docofossor 160 Mya 0 brachydactylus (Luo, Meng, Ji et al., 2015) Docodonta Haldanodon 150-155 Mya, Late 0 Shallow patellar groove exspectatus Jurassic, Portugal (Martin, 2005b) Australosphenida Asfaltomylos Mid-Jurassic, South ? Postcranial material absent patagonicus America (Martin, 2005a) Australosphenida Ornithorhynchus Extant 2 Platypus, genome sequenced Monotremata anatinus (Warren, Hillier, Marshall Graves et (Herzmark, 1938; al., 2008) Rowe, 1988) Samuels et al. Australosphenida Tachyglossus + Extant 2 Echidnas Monotremata Zaglossus spp. (Herzmark, 1938; Rowe, 1988) Mammaliaformes Fruitafossor 150 Mya, Late Jurassic, 0 Phylogenetic status uncertain indet. windscheffeli (Luo Colorado & Wible, 2005) Mammaliaformes Volaticotherium Late Jurassic/Early ? Hindlimb material incomplete indet. antiquus (Meng, Cretaceous Hu, Wang et al., 2006) Eutriconodonta Jeholodens 120-125 Mya, Early 0 Poorly developed patellar groove jenkinsi (Ji, Luo Cretaceous, China & Ji, 1999) Eutriconodonta Gobiconodon spp. -

To Link and Cite This Article: Doi
Submitted: October 1st, 2019 – Accepted: June 8th, 2020 – Published online: June 11th, 2020 To link and cite this article: doi: https://doi.org/10.5710/AMGH.08.06.2020.3306 1 COTYLAR FOSSA, ITS INTERPRETATION AND FUNCTIONALITY. 2 THE CASE FROM SOUTH AMERICAN NATIVE UNGULATES 3 4 MALENA LORENTE1,2, 3 5 1 División Paleontología de Vertebrados, Museo de La Plata. Paseo del Bosque s/n B1900FWA, La 6 Plata, Buenos Aires, Argentina; [email protected]; 2 CONICET; 3 Facultad de Ciencias 7 Naturales y Museo 8 9 Number of pages: 21, 7 figures 10 Proposed header: LORENTE, COTYLAR FOSSA 1 11 12 Abstract. The cotylar fossa is a complex anatomical character in the astragalar medial malleolar 13 facet. It represents a dynamic relationship between the astragalus and the tibia in the upper tarsal 14 joint. The astragalus must accommodate the medial tibial malleolus when the tibia is in an extreme 15 flexion. It is one of the three morphological synapomorphies considered for Afrotheria, although it 16 is also a recurrent trait among different groups of mammals. Here, fossil South American Native 17 Ungulates and extant mammals were surveyed to reevaluate how much this character is spread and 18 how variable it is. Beyond afrotherians, it is observed that it also appears in primates, macropodid 19 marsupials, laurasiatherian archaic ungulates, perissodactyls, pantodonts, and dinoceratans; it is also 20 in some but not all of the extinct endemic ungulates from South America. No function has been 21 suggested before for the presence of a cotylar fossa. The cotylar fossa could be an adaptation to a 22 passive, rest related posture. -

Afrotheria Slide Show
Afrotheria Specialist Group Afrotheria SPECIALIST GROUP 3) 2) 1) Afrotheria SpecialistfacilitatesGroupIUCN/SSC The programmes. developingconservation researchand and publicraisingawareness; interested groups; otherand conservationists,governments, providingscientific sound guidance toadviceand golden-moles,theirtenrecs,habitatsand by: sengis, aardvark, the hyraxes, the of conservation The AfrotheriaIUCN/SSC Specialist Group © Stephanie Mansourian P U The Afrotheria Specialist Group Is Concerned With 76 Species O a R i Of Mammals From Africa And Madagascar G r T S I e L A I C h E t P S o r © S. Cilliers f © Peter J. Stephenson 1 Aardvark A 33 Tenrecs © H. Hoeck 5 Hyraxes © G.B. Rathbun © G.B. Rathbun 16 Sengis 21 Golden Moles One quarter are Threatened, and several others are NearThreatened or too Data Deficient to categorize Afrotheria SPECIALIST GROUP • • • and distribution of its ant and termite prey. and might be definedby the abundance Aardvark distribution is not well known up to Weighs 60 kgand occursacross mostof Africa south of the Sahara. evolution. South America, but this onlywas based on superficial resemblancesdue to convergent The last used it living species to be ofconsidered its Order, a relativeof anteaters from Aardvark (Tubulidentata) Aardvark © T. Lehmann Afrotheria SPECIALIST GROUP • • • • • • • • • 10golden mole species are greatestThe goldendiversity of moles Africa.southernisin smallthese mammalsblind,issubterranean stilllimited. firstLinnaeusdescribe goldenknowledge yet of moles ago, 250nearly years forests. managementofindigenous development,orpoor urbanization, agricultural degradationfrom mining, threateneddue tohabitat GoldenMoles (Chrysochloridae) © G.Bronner Afrotheria SPECIALIST GROUP • • • mostofallrisk at hyraxes. are probably TreeHyraxes the alarmingthe rateanat Africa, in forests Because are disappearing Kilimanjaro.Mt. around the inforestbelt itsforfur hunted heavilyis TreeHyrax Eastern The seacows,otherelephantstheand afrotheriantwo taxa.