1W25 Lichtarge Lab 2006
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Studying GGDEF Domain in the Act: Minimize Conformational Frustration to Prevent Artefacts
life Article Studying GGDEF Domain in the Act: Minimize Conformational Frustration to Prevent Artefacts Federico Mantoni 1,†, Chiara Scribani Rossi 1,2,†, Alessandro Paiardini 1,2, Adele Di Matteo 3 , Loredana Cappellacci 4 , Riccardo Petrelli 4 , Massimo Ricciutelli 4, Alessio Paone 1,2, Francesca Cutruzzolà 1,2, Giorgio Giardina 1,2,* and Serena Rinaldo 1,2,* 1 Department of Biochemical Sciences “A. Rossi Fanelli”, Sapienza University of Rome, Piazzale Aldo Moro, 5, 00185 Rome, Italy; [email protected] (F.M.); [email protected] (C.S.R.); [email protected] (A.P.); [email protected] (A.P.); [email protected] (F.C.) 2 Laboratory Affiliated to Istituto Pasteur Italia—Fondazione Cenci Bolognetti, 00185 Rome, Italy 3 Institute of Molecular Biology and Pathology, CNR, Piazzale Aldo Moro, 5, 00185 Rome, Italy; [email protected] 4 Medicinal Chemistry Unit, School of Pharmacy, University of Camerino, Via S. Agostino 1, 62032 Camerino, Italy; [email protected] (L.C.); [email protected] (R.P.); [email protected] (M.R.) * Correspondence: [email protected] (G.G.); [email protected] (S.R.) † These authors contributed equally to the work. Abstract: GGDEF-containing proteins respond to different environmental cues to finely modulate cyclic diguanylate (c-di-GMP) levels in time and space, making the allosteric control a distinctive trait of the corresponding proteins. The diguanylate cyclase mechanism is emblematic of this control: two GGDEF domains, each binding one GTP molecule, must dimerize to enter catalysis and yield c-di-GMP. The need for dimerization makes the GGDEF domain an ideal conformational switch in Citation: Mantoni, F.; Scribani Rossi, multidomain proteins. -

Supplemental Table 7. Every Significant Association
Supplemental Table 7. Every significant association between an individual covariate and functional group (assigned to the KO level) as determined by CPGLM regression analysis. Variable Unit RelationshipLabel See also CBCL Aggressive Behavior K05914 + CBCL Emotionally Reactive K05914 + CBCL Externalizing Behavior K05914 + K15665 K15658 CBCL Total K05914 + K15660 K16130 KO: E1.13.12.7; photinus-luciferin 4-monooxygenase (ATP-hydrolysing) [EC:1.13.12.7] :: PFAMS: AMP-binding enzyme; CBQ Inhibitory Control K05914 - K12239 K16120 Condensation domain; Methyltransferase domain; Thioesterase domain; AMP-binding enzyme C-terminal domain LEC Family Separation/Social Services K05914 + K16129 K16416 LEC Poverty Related Events K05914 + K16124 LEC Total K05914 + LEC Turmoil K05914 + CBCL Aggressive Behavior K15665 + CBCL Anxious Depressed K15665 + CBCL Emotionally Reactive K15665 + K05914 K15658 CBCL Externalizing Behavior K15665 + K15660 K16130 KO: K15665, ppsB, fenD; fengycin family lipopeptide synthetase B :: PFAMS: Condensation domain; AMP-binding enzyme; CBCL Total K15665 + K12239 K16120 Phosphopantetheine attachment site; AMP-binding enzyme C-terminal domain; Transferase family CBQ Inhibitory Control K15665 - K16129 K16416 LEC Poverty Related Events K15665 + K16124 LEC Total K15665 + LEC Turmoil K15665 + CBCL Aggressive Behavior K11903 + CBCL Anxiety Problems K11903 + CBCL Anxious Depressed K11903 + CBCL Depressive Problems K11903 + LEC Turmoil K11903 + MODS: Type VI secretion system K01220 K01058 CBCL Anxiety Problems K11906 + CBCL Depressive -

The Degenerate EAL-GGDEF Domain Protein Filp Functions As a Cyclic
MPMI Vol. 27, No. 6, 2014, pp. 578–589. http://dx.doi.org/10.1094/MPMI-12-13-0371-R e-Xtra* The Degenerate EAL-GGDEF Domain Protein Filp Functions as a Cyclic di-GMP Receptor and Specifically Interacts with the PilZ-Domain Protein PXO_02715 to Regulate Virulence in Xanthomonas oryzae pv. oryzae Fenghuan Yang,1 Fang Tian,1 Xiaotong Li,1 Susu Fan,1 Huamin Chen,1 Maosen Wu,1 Ching-Hong Yang,2 and Chenyang He1 1State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing 100193; 2Department of Biological Sciences, University of Wisconsin-Milwaukee, Milwaukee 53211, U.S.A. Submitted 18 December 2013. Accepted 8 February 2014. Degenerate GGDEF and EAL domain proteins represent the production of exopolysaccharides (EPS) and extracellular major types of cyclic diguanylic acid (c-di-GMP) receptors enzymes, the expression of type III secretion system genes, in pathogenic bacteria. Here, we characterized a FimX-like and so on (Fouhy et al. 2006; Kalia et al. 2013; Kulasakara et protein (Filp) which possesses both GGDEF and EAL al. 2006; Yi et al. 2010). Synthesis and degradation of c-di- domains in Xanthomonas oryzae pv. oryzae, the causal GMP is controlled by the opposing activities of diguanylate agent of bacterial blight of rice. Both in silico analysis and cyclases (DGC), which contain the GGDEF domain, and phos- enzyme assays indicated that the GGDEF and EAL do- phodiesterases (PDE), which harbor EAL or HD-GYP domains mains of Filp were degenerate and enzymatically inactive. (Schirmer and Jenal 2009). -

The Diversity of Globin-Coupled Sensors
FEBS 27611 FEBS Letters 552 (2003) 99^104 View metadata, citation and similar papers at core.ac.uk brought to you by CORE Minireview provided by Elsevier - Publisher Connector The diversity of globin-coupled sensors Tracey Allen K. Freitasa;b, Shaobin Houa, Maqsudul Alama;b;Ã aDepartment of Microbiology, Snyder Hall 207, 2538 The Mall, University of Hawaii, Honolulu, HI 96822, USA bMaui High Performance Computing Center, 550 Lipoa Parkway, Kihei, Maui, HI 96753, USA Received 2 July 2003; revised 5 August 2003; accepted 10 August 2003 First published online 5 September 2003 Edited by Horst Feldmann cGMP 2nd messenger. The direct oxygen sensor (Dos), ¢rst Abstract The recently discovered globin-coupled sensors (GCSs) are heme-containing two-domain transducers distinct described in Escherichia coli [8], functions as a tetrameric from the PAS domain superfamily. We have identi¢ed an addi- phosphodiesterase (PDE) by converting cAMP to 5P-AMP tional 22 GCSs with varying multi-domain C-terminal transmit- while in the ferrous form, and is strongly inhibited by CO ters through a search of the complete and incomplete microbial and NO ligands [9]. A1 from Acetobacter xylinum (AxP- genome datasets. The GCS superfamily is composed of two DEA1) also functions as a PDE by linearizing cyclic major subfamilies: the aerotactic and gene regulators. We pos- bis(3PC5P)diguanylate, an allosteric activator of the bacterial tulate the existence of protoglobin in Archaea as the predecessor cellulose synthase, to the ine¡ectual pGpG [10,11]. Both Dos to the chimeric GCS. and AxPDEA1 possess similar heme-binding PAS domains ß 2003 Published by Elsevier B.V. -

Us 2018 / 0237793 A1
US 20180237793A1 ( 19) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0237793 A1 Aasen et al. (43 ) Pub . Date : Aug. 23, 2018 ( 54 ) TRANSGENIC PLANTS WITH ENHANCED Tyamagondlu V . Venkatesh , St . Louis , AGRONOMIC TRAITS MO (US ) ; Huai Wang, Chesterfield , MO (US ) ; Jingrui Wu , Johnston , IA (71 ) Applicant: Monsanto Technology LLC , St. Louis , (US ) ; Xiaoyun Wu, Chesterfield , MO MO (US ) (US ) ; Xiaofeng Sean Yang , Chesterfield , MO (US ) ; Qin Zeng , ( 72 ) Inventors : Eric D . Aasen , Woodland , CA (US ) ; Maryland Heights , MO (US ) ; Jianmin Mark Scott Abad , Webster Groves , Zhao , St . Louis , MO (US ) MO (US ) ; Edwards Allen , O ' Fallon , MO (US ) ; Thandoni Rao Ambika , (73 ) Assignee: Monsanto Technology LLC , St. Louis , Karnataka (IN ) ; Veena S . Anil , MO (US ) Bangalore ( IN ) ; Alice Clara Augustine , (21 ) Appl. No .: 15 /827 , 886 Karnataka ( IN ) ; Stephanie L . Back , St. Louis , MO (US ) ; Pranesh Badami, (22 ) Filed : Nov . 30 , 2017 Bangalore (IN ) ; Amarjit Basra , Chesterfield , MO (US ) ; Kraig L . Related U . S . Application Data Brustad , Saunderstown , RI (US ) ; (63 ) Continuation of application No . 14 /632 , 018 , filed on Molian Deng, Grover , MO (US ) ; Todd Feb . 26 , 2015 , now abandoned , which is a continu Dezwaan , Apex , NC (US ) ; Charles ation of application No. 12 /998 ,243 , filed on Sep . 16 , Dietrich , Chesterfield , MO (US ) ; 2011 , now abandoned , filed as application No . PCT/ Stephen M . Duff , St. Louis , MO (US ) ; US2009/ 058908 on Sep . 30 , 2009 . Karen K . Gabbert , St. Louis , MO ( 60 ) Provisional application No . 61/ 226 , 953 , filed on Jul . ( US ) ; Steve He, St. Louis , MO (US ) ; 20 , 2009 , provisional application No . 61 / 182 ,785 , Bill L . -

The Response of Cupriavidus Metallidurans CH34 to Cadmium
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 26 July 2018 doi:10.20944/preprints201807.0520.v1 Article The Response of Cupriavidus Metallidurans CH34 to Cadmium Involves a Decrease in Intracellular Levels of C-Di-GMP Probably Mediated by a Novel Metal Regulated Phosphodiesterase That Inhibits the Biofilm Lifestyle Pablo Alviz-Gazitua 1,2,3, Sebastián Fuentes 4, Luis Rojas 2,5, Raymond J. Turner 6, Michael Seeger 2,* and Nicolás Guiliani 1,* 1. Bacterial Communication Laboratory, Biology Department, Faculty of Sciences, Universidad de Chile, Ñuñoa, Santiago, Chile. [email protected] 2. Molecular Microbiology and Environmental Biotechnology Laboratory, Chemistry Department & Center of Biotechnology, Universidad Técnica Federico Santa María, Valparaíso, Chile. 3. PhD program of Microbiology, Faculty of Sciences, Universidad de Chile, Ñuñoa, Santiago, Chile. 4. Microbial Ecology of Extreme Systems Laboratory, Biological Sciences Faculty, Pontificia Universidad Católica de Chile, Santiago, Chile. 5. Chemistry Department, Universidad Católica del Norte, Avenida Angamos 0610, Antofagasta CL- 1240000, Chile 6. Biofilm Research Group, Department of Biological Sciences, University of Calgary, Calgary, Canada. * Correspondence: [email protected], [email protected]; Tel.: +56-322654223 & [email protected]; Tel.: +56-229787241. These authors contributed equally to this work. Abstract: Cadmium is a highly toxic heavy metal for biological systems. Cupriavidus metallidurans CH34 is a model strain for heavy metal resistance and bioremediation. The aim of this study was to determine the role of the c-di-GMP pathway in the C. metallidurans CH34 response to cadmium in both planktonic and biofilm cells. Increasing cadmium concentrations correlates with an inhibition of biofilm formation and EPS production in C. -

Hybrid Promiscuous (Hypr) GGDEF Enzymes Produce Cyclic AMP-GMP (3′, 3′-Cgamp)
Hybrid promiscuous (Hypr) GGDEF enzymes produce cyclic AMP-GMP (3′,3′-cGAMP) Zachary F. Hallberga, Xin C. Wangb, Todd A. Wrighta, Beiyan Nanc, Omer Ada, Jongchan Yeoa, and Ming C. Hammonda,b,1 aDepartment of Chemistry, University of California, Berkeley, CA 94720; bDepartment of Molecular and Cell Biology, University of California, Berkeley, CA 94720; and cDepartment of Biology, Texas A&M University, College Station, TX 77843 Edited by Bonnie L. Bassler, Princeton University and Howard Hughes Medical Institute, Princeton, NJ, and approved December 30, 2015 (received for review August 3, 2015) Over 30 years ago, GGDEF domain-containing enzymes were shown or cGAS, which harbor oligoadenylate synthase (OAS)-like do- to be diguanylate cyclases that produce cyclic di-GMP (cdiG), a second mains and produce structurally distinct isomers of cAG (3′,3′- messenger that modulates the key bacterial lifestyle transition from a cGAMP and 2′,3′-cGAMP, respectively) (12–14, 20). However, motile to sessile biofilm-forming state. Since then, the ubiquity of the cAG-sensing riboswitches in Geobacter gained function genes encoding GGDEF proteins in bacterial genomes has established via adapting the ligand binding pocket of GEMM-I riboswitches, the dominance of cdiG signaling in bacteria. However, the observa- which typically bind cdiG (21). Thus, we considered that cAG tion that proteobacteria encode a large number of GGDEF proteins, signaling may have evolved in Geobacter by co-opting components nearing 1% of coding sequences in some cases, raises the question of from the cdiG signaling pathway. why bacteria need so many GGDEF enzymes. In this study, we reveal The G. -

STING Cyclic Dinucleotide Sensing Originated in Bacteria
Article STING cyclic dinucleotide sensing originated in bacteria https://doi.org/10.1038/s41586-020-2719-5 Benjamin R. Morehouse1,2, Apurva A. Govande1,2, Adi Millman3, Alexander F. A. Keszei4, Brianna Lowey1,2, Gal Ofir3, Sichen Shao4, Rotem Sorek3 & Philip J. Kranzusch1,2,5 ✉ Received: 2 May 2020 Accepted: 26 August 2020 Stimulator of interferon genes (STING) is a receptor in human cells that senses foreign Published online: 2 September 2020 cyclic dinucleotides that are released during bacterial infection and in endogenous Check for updates cyclic GMP–AMP signalling during viral infection and anti-tumour immunity1–5. STING shares no structural homology with other known signalling proteins6–9, which has limited attempts at functional analysis and prevented explanation of the origin of cyclic dinucleotide signalling in mammalian innate immunity. Here we reveal functional STING homologues encoded within prokaryotic defence islands, as well as a conserved mechanism of signal activation. Crystal structures of bacterial STING defne a minimal homodimeric scafold that selectively responds to cyclic di-GMP synthesized by a neighbouring cGAS/DncV-like nucleotidyltransferase (CD-NTase) enzyme. Bacterial STING domains couple the recognition of cyclic dinucleotides with the formation of protein flaments to drive oligomerization of TIR efector domains and rapid NAD+ cleavage. We reconstruct the evolutionary events that followed the acquisition of STING into metazoan innate immunity, and determine the structure of a full-length TIR–STING fusion from the Pacifc oyster Crassostrea gigas. Comparative structural analysis demonstrates how metazoan-specifc additions to the core STING scafold enabled a switch from direct efector function to regulation of antiviral transcription. -
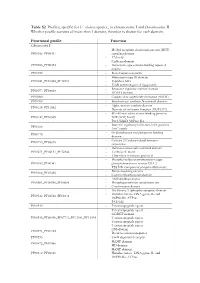
Table S2. Profiles, Specific for V. Cholerae Species., in Chromosome I and Chromosome II
Table S2. Profiles, specific for V. cholerae species., in chromosome I and chromosome II. Whether profile consists of more than 1 domain, function is shown for each domain. Functional profile Function Chromosome I - Methyl-accepting chemotaxis protein (MCP) PF00015_PF08447 signalling domain - PAS fold - Cadherin domain PF00028_PF00353 - Hemolysin-type calcium-binding repeat (2 copies) PF00030 - Beta/Gamma crystallin - Fibronectin type III domain PF00041_PF10462_PF12561 - Peptidase M66 - ToxR activated gene A lipoprotein - Response regulator receiver domain PF00072_PF08668 - HDOD domain PF00080 - Copper/zinc superoxide dismutase (SODC) PF00109 - Beta-ketoacyl synthase N-terminal domain - Alpha amylase catalytic domain PF00128_PF11852 - Domain of unknown function (DUF3372) - 4Fe-4S iron sulfur cluster binding proteins PF00142_PF10609 NifH/frxC family - ParA/MinD ATPase like - Bacterial regulatory helix-turn-helix proteins PF00165 AraC family - Oxidoreductase molybdopterin binding PF00174 domain - Catalase||Catalase-related immune- PF00199_PF06628 responsive - Helicase conserved C-terminal domain PF00271_PF02151_PF12344 - UvrB/uvrC motif - Ultra-violet resistance protein B - Phosphoenolpyruvate-dependent sugar PF00359_PF00381 phosphotransferase system EIIA 2 - PTS HPr component phosphorylation site - Biotin-requiring enzyme PF00364_PF02436 - Conserved carboxylase domain - AMP-binding enzyme PF00501_PF00550_PF00668 - Phosphopantetheine attachment site - Condensation domain - His Kinase A (phospho-acceptor) domain - Histidine kinase- DNA gyrase -

2 12/ 1696 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date k i ft - 9 February 2012 (09.02.2012) 2 12/ 1696 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C07K 14/365 (2006.01) C12P 19/26 (2006.01) kind of national protection available): AE, AG, AL, AM, C12N 15/11 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (21) Number: International Application DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/EP201 1/063243 HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (22) International Filing Date: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, 1 August 201 1 (01 .08.201 1) ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (25) Filing Language: English SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, (26) Publication Language: English TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 1017 183 1.0 4 August 2010 (04.08.2010) EP kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, (71) Applicant (for all designated States except US): BAYER ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, PHARMA AKTIENGESELLSCHAFT [DE/DE]; TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Muller Strasse 178, 13353 Berlin (DE). -

A Cyclic-Di-GMP Signalling Network Regulates Biofilm Formation And
http://www.diva-portal.org This is the published version of a paper published in Scientific Reports. Citation for the original published paper (version of record): Ahmad, I., Nygren, E., Khalid, F., Myint, S L., Uhlin, B E. (2020) A Cyclic-di-GMP signalling network regulates biofilm formation and surface associated motility of Acinetobacter baumannii 17978 Scientific Reports, 10(1): 1991 https://doi.org/10.1038/s41598-020-58522-5 Access to the published version may require subscription. N.B. When citing this work, cite the original published paper. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:umu:diva-170711 www.nature.com/scientificreports OPEN A Cyclic-di-GMP signalling network regulates bioflm formation and surface associated motility of Acinetobacter baumannii 17978 Irfan Ahmad1,2*, Evelina Nygren 1, Fizza Khalid2, Si Lhyam Myint1 & Bernt Eric Uhlin 1* Acinetobacter baumannii has emerged as an increasing multidrug-resistant threat in hospitals and a common opportunistic nosocomial pathogen worldwide. However, molecular details of the pathogenesis and physiology of this bacterium largely remain to be elucidated. Here we identify and characterize the c-di-GMP signalling network and assess its role in bioflm formation and surface associated motility. Bioinformatic analysis revealed eleven candidate genes for c-di-GMP metabolizing proteins (GGDEF/EAL domain proteins) in the genome of A. baumannii strain 17978. Enzymatic activity of the encoded proteins was assessed by molecular cloning and expression in the model organisms Salmonella typhimurium and Vibrio cholerae. Ten of the eleven GGDEF/EAL proteins altered the rdar morphotype of S. typhimurium and the rugose morphotype of V. -

Genes Encoding Cher-TPR Fusion Proteins Are Predominantly Found in Gene Clusters Encoding Chemosensory Pathways with Alternative Cellular Functions
Genes Encoding Cher-TPR Fusion Proteins Are Predominantly Found in Gene Clusters Encoding Chemosensory Pathways with Alternative Cellular Functions Francisco Mun˜ oz-Martı´nez1., Cristina Garcı´a-Fontana1., Miriam Rico-Jime´nez1, Carlos Alfonso2, Tino Krell1* 1 Department of Environmental Protection, Estacio´n Experimental del Zaidı´n, Consejo Superior de Investigaciones Cientı´ficas, Granada, Spain, 2 Centro de Investigaciones Biolo´gicas, Madrid, Spain Abstract Chemosensory pathways correspond to major signal transduction mechanisms and can be classified into the functional families flagellum-mediated taxis, type four pili-mediated taxis or pathways with alternative cellular functions (ACF). CheR methyltransferases are core enzymes in all of these families. CheR proteins fused to tetratricopeptide repeat (TPR) domains have been reported and we present an analysis of this uncharacterized family. We show that CheR-TPRs are widely distributed in GRAM-negative but almost absent from GRAM-positive bacteria. Most strains contain a single CheR-TPR and its abundance does not correlate with the number of chemoreceptors. The TPR domain fused to CheR is comparatively short and frequently composed of 2 repeats. The majority of CheR-TPR genes were found in gene clusters that harbor multidomain response regulators in which the REC domain is fused to different output domains like HK, GGDEF, EAL, HPT, AAA, PAS, GAF, additional REC, HTH, phosphatase or combinations thereof. The response regulator architectures coincide with those reported for the ACF family of pathways. Since the presence of multidomain response regulators is a distinctive feature of this pathway family, we conclude that CheR-TPR proteins form part of ACF type pathways. The diversity of response regulator output domains suggests that the ACF pathways form a superfamily which regroups many different regulatory mechanisms, in which all CheR-TPR proteins appear to participate.