Stereochemistry of Alkanes and Cycloalkanes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Prebiological Evolution and the Metabolic Origins of Life
Prebiological Evolution and the Andrew J. Pratt* Metabolic Origins of Life University of Canterbury Keywords Abiogenesis, origin of life, metabolism, hydrothermal, iron Abstract The chemoton model of cells posits three subsystems: metabolism, compartmentalization, and information. A specific model for the prebiological evolution of a reproducing system with rudimentary versions of these three interdependent subsystems is presented. This is based on the initial emergence and reproduction of autocatalytic networks in hydrothermal microcompartments containing iron sulfide. The driving force for life was catalysis of the dissipation of the intrinsic redox gradient of the planet. The codependence of life on iron and phosphate provides chemical constraints on the ordering of prebiological evolution. The initial protometabolism was based on positive feedback loops associated with in situ carbon fixation in which the initial protometabolites modified the catalytic capacity and mobility of metal-based catalysts, especially iron-sulfur centers. A number of selection mechanisms, including catalytic efficiency and specificity, hydrolytic stability, and selective solubilization, are proposed as key determinants for autocatalytic reproduction exploited in protometabolic evolution. This evolutionary process led from autocatalytic networks within preexisting compartments to discrete, reproducing, mobile vesicular protocells with the capacity to use soluble sugar phosphates and hence the opportunity to develop nucleic acids. Fidelity of information transfer in the reproduction of these increasingly complex autocatalytic networks is a key selection pressure in prebiological evolution that eventually leads to the selection of nucleic acids as a digital information subsystem and hence the emergence of fully functional chemotons capable of Darwinian evolution. 1 Introduction: Chemoton Subsystems and Evolutionary Pathways Living cells are autocatalytic entities that harness redox energy via the selective catalysis of biochemical transformations. -

Chapter 3 • Ratio: Cnh2n+2
Organic Chemistry, 5th Edition Alkane Formulas L. G. Wade, Jr. • All C-C single bonds • Saturated with hydrogens Chapter 3 • Ratio: CnH2n+2 Structure and Stereochemistry • Alkane homologs: CH3(CH2)nCH3 of Alkanes • Same ratio for branched alkanes H H H H H H CH H C C C C H H H H C CCH Jo Blackburn H H H H H H Richland College, Dallas, TX H => Butane, C4H10 Dallas County Community College District Chapter 3Isobutane, C H 2 © 2003, Prentice Hall 4 10 Common Names Pentanes H H H H H H H CH • Isobutane, “isomer of butane” H C C C C C H H H • Isopentane, isohexane, etc., methyl H H H H H H H C CCC H branch on next-to-last carbon in chain. H H H H • Neopentane, most highly branched n-pentane, C5H12 isopentane, C5H12 • Five possible isomers of hexane, CH3 18 isomers of octane and 75 for H3C C CH3 decane! CH3 => => neopentane, C5H12 Chapter 3 3 Chapter 3 4 Longest Chain IUPAC Names • The number of carbons in the longest • Find the longest continuous carbon chain determines the base name: chain. ethane, hexane. (Listed in Table 3.2, • Number the carbons, starting closest to page 81.) the first branch. • If there are two possible chains with the • Name the groups attached to the chain, same number of carbons, use the chain using the carbon number as the locator. with the most substituents. • Alphabetize substituents. H3C CH CH CH • Use di-, tri-, etc., for multiples of same 2 3 CH3 substituent. -

Chapter 4: Stereochemistry Introduction to Stereochemistry
Chapter 4: Stereochemistry Introduction To Stereochemistry Consider two of the compounds we produced while finding all the isomers of C7H16: CH3 CH3 2-methylhexane 3-methylhexane Me Me Me C Me H Bu Bu Me Me 2-methylhexane H H mirror Me rotate Bu Me H 2-methylhexame is superimposable with its mirror image Introduction To Stereochemistry Consider two of the compounds we produced while finding all the isomers of C7H16: CH3 CH3 2-methylhexane 3-methylhexane H C Et Et Me Pr Pr 3-methylhexane Me Me H H mirror Et rotate H Me Pr 2-methylhexame is superimposable with its mirror image Introduction To Stereochemistry Consider two of the compounds we produced while finding all the isomers of C7H16: CH3 CH3 2-methylhexane 3-methylhexane .Compounds that are not superimposable with their mirror image are called chiral (in Greek, chiral means "handed") 3-methylhexane is a chiral molecule. .Compounds that are superimposable with their mirror image are called achiral. 2-methylhexane is an achiral molecule. .An atom (usually carbon) with 4 different substituents is called a stereogenic center or stereocenter. Enantiomers Et Et Pr Pr Me CH3 Me H H 3-methylhexane mirror enantiomers Et Et Pr Pr Me Me Me H H Me H H Two compounds that are non-superimposable mirror images (the two "hands") are called enantiomers. Introduction To Stereochemistry Structural (constitutional) Isomers - Compounds of the same molecular formula with different connectivity (structure, constitution) 2-methylpentane 3-methylpentane Conformational Isomers - Compounds of the same structure that differ in rotation around one or more single bonds Me Me H H H Me H H H H Me H Configurational Isomers or Stereoisomers - Compounds of the same structure that differ in one or more aspects of stereochemistry (how groups are oriented in space - enantiomers or diastereomers) We need a a way to describe the stereochemistry! Me H H Me 3-methylhexane 3-methylhexane The CIP System Revisited 1. -

Organic Chemistry by Robert C
(2/94)(6,7,9/95)(8,9/97)(12/99)(1/00) Neuman Chapter 4 Chapter 4 Stereochemistry from Organic Chemistry by Robert C. Neuman, Jr. Professor of Chemistry, emeritus University of California, Riverside [email protected] <http://web.chem.ucsb.edu/~neuman/orgchembyneuman/> Chapter Outline of the Book ************************************************************************************** I. Foundations 1. Organic Molecules and Chemical Bonding 2. Alkanes and Cycloalkanes 3. Haloalkanes, Alcohols, Ethers, and Amines 4. Stereochemistry 5. Organic Spectrometry II. Reactions, Mechanisms, Multiple Bonds 6. Organic Reactions *(Not yet Posted) 7. Reactions of Haloalkanes, Alcohols, and Amines. Nucleophilic Substitution 8. Alkenes and Alkynes 9. Formation of Alkenes and Alkynes. Elimination Reactions 10. Alkenes and Alkynes. Addition Reactions 11. Free Radical Addition and Substitution Reactions III. Conjugation, Electronic Effects, Carbonyl Groups 12. Conjugated and Aromatic Molecules 13. Carbonyl Compounds. Ketones, Aldehydes, and Carboxylic Acids 14. Substituent Effects 15. Carbonyl Compounds. Esters, Amides, and Related Molecules IV. Carbonyl and Pericyclic Reactions and Mechanisms 16. Carbonyl Compounds. Addition and Substitution Reactions 17. Oxidation and Reduction Reactions 18. Reactions of Enolate Ions and Enols 19. Cyclization and Pericyclic Reactions *(Not yet Posted) V. Bioorganic Compounds 20. Carbohydrates 21. Lipids 22. Peptides, Proteins, and α−Amino Acids 23. Nucleic Acids ************************************************************************************** -

Stereochemistry of Bruice's Organic Chemistry Before Embarking on Comprehending This Summary
Enantioselective synthesis It helps if you review the concepts in Chapter 5: Stereochemistry of Bruice's Organic Chemistry before embarking on comprehending this summary Most of the molecules on earth are chiral, such as amino acids and carbohydrates. Enantioselective synthesis is an important means by which enantiopure chiral molecules may be obtained for study and medicine. The study of enantioselective synthesis has evolved from issues of diastereoselectivity, through auxiliary-based methods for the synthesis of enantiomerically pure compounds (diastereoselectivity followed by separation and auxiliary cleavage), to asymmetric catalysis. In the latter technology, enantiomers are the products, and highly selective reactions allow preparation - in a single step - of chiral substances in high enantiomeric excess (ee) for many reaction types. Enantioselective synthesis, also known as asymmetric synthesis, is organic synthesis that introduces one or more new and desired elements of chirality. This methodology is important in the field of pharmaceuticals because the different enantiomers or diastereomers of a molecule often have different biological activity. See examples from Chapter 5 of Bruice's Organic Chemistry. Methodologies of Asymmetric Synthesis Chiral pool synthesis Chiral pool synthesis is the easiest approach: A chiral starting material is manipulated through successive reactions using achiral reagents that retain its chirality to obtain the desired target molecule. This is especially attractive for target molecules having the similar chirality to a relatively inexpensive naturally occurring building-block such as a sugar or amino acid. However, the number of possible reactions the molecule can undergo is restricted, and lengthy synthetic routes may be required. Also, this approach requires a stoichiometric amount of the enantiopure starting material, which may be rather expensive if not occurring in nature, whereas chiral catalysis requires only a catalytic amount of chiral material. -

NMR and Stereochemistry Chem 4010/5326: Organic Spectroscopic Analysis
NMR and Stereochemistry Chem 4010/5326: Organic Spectroscopic Analysis © 2015 Andrew Harned General flow for solving structures C10H20O Molecular weight/formula (MS) Exact Mass: 156.1514 Molecular Weight: 156.2652 Functional groups (IR, NMR) Carbon connectivities (substructures) (NMR) Positions of functional groups within framework (gross structure) (2D NMR, coupling constants) How can this Stereochemical issues be solved??? Relative Stereochemistry (Diastereomers) Can be determined with many of the tools we have already discussed, along with some new ones Bifulco, G.; Dambruoso, P.; Gomex-Paloma, L.; Riccio, R. Chem. Rev. 2007, 107, 3744. NMR Spectroscopy Strategies for determining relative stereochemistry Chemical Shifts – Diastereotopic protons will have different chemical shifts, this will only tell you that diastereomers are present, cannot necessarily tell which is which by inspection only by comparison to known structures – Spatial orientation may place certain protons in shielding/deshielding portions of functional groups Coupling Constants – In acyclic systems, usually cannot tell which is which by inspection – Often must convert to rigid/cyclic structure Both require some knowledge of 3D structure –> Make model(s) NMR Spectroscopy Proximity of Protons Nuclear Overhauser Effect (nOe) – Through space interactions between nuclei, whether or not they are directly coupled – Magnitude decreases as inverse of sixth power of distance – Strongly irradiate one, get larger # in excited state – The others then shift to lower state to compensate -

Asymmetric Synthesis
Chapter 45 — Asymmetric synthesis - Pure enantiomers from Nature: the chiral pool and chiral induction - Asymmetric synthesis: chiral auxiliaries Enolate alkylation Aldol reaction - Enantiomeric excess (ee) - Asymmetric synthesis: chiral reagents and catalysts CBS reagent for chiral reductions Sharpless asymmetric epoxidation Synthesizing pure enantiomers starting from Nature’s chiral pool AcO OH HO B O O HO – O O HO + O N OH H3N OH HO OH O H … AcO HO OH O OH Sulcatol - insect pheremone HO Synthesis from the chiral pool — chiral induction turns one stereocenter into many 40 steps Me HO H Me H O O OH O O OBn O * * Only one H O HO chiral reagent H H Me OBn Me Deoxyribose Fragment of Brevetoxin B Asymmetric synthesis 1. Producing a new stereogenic centre on an achiral molecule makes two enantiomeric transition states of equal energy… and therefore two enantiomeric products in equal amounts. O O 1 1 Nu R R2 R R2 Nu E OH OH 1 O 1 Nu R R Nu R2 R2 1 R R2 O Ph OH HO Ph PhLi + N N N Asymmetric synthesis 2. O 1 R R* Nu When there is an existing O chiral centre, the two possible TS’s are diastereomeric and E 1 Nu R R* can be of different energy. Thus one isomer of the new stereogenic centre can be OH O OH produced in a larger amount. 1 1 R Nu Nu R *R 1 R* R R* HO Ph O O O Ph OH PhLi PhLi Ph Ph N N N N N Ph N N N N N OH Ph O O O Ph OH Ph A removable chiral centre… synthesis with chiral auxiliaries O ??? O R R 1) Add a chiral 3) Remove the auxiliary chiral auxiliary O O R* R* 2) Add the new stereocentre via chiral induction Enantiopure oxazolidinones as chiral auxiliaries 1. -

Reactions of Alkenes Since Bonds Are Stronger Than Bonds, Double Bonds Tend to React to Convert the Double Bond Into Bonds
Reactions of Alkenes Since bonds are stronger than bonds, double bonds tend to react to convert the double bond into bonds This is an addition reaction. (Other types of reaction have been substitution and elimination). Addition reactions are typically exothermic. Electrophilic Addition The bond is localized above and below the C-C bond. The electrons are relatively far away from the nuclei and are therefore loosely bound. An electrophile will attract those electrons, and can pull them away to form a new bond. This leaves one carbon with only 3 bonds and a +ve charge (carbocation). The double bond acts as a nucleophile (attacks the electrophile). Ch08 Reacns of Alkenes (landscape) Page 1 In most cases, the cation produced will react with another nucleophile to produce the final overall electrophilic addition product. Electrophilic addition is probably the most common reaction of alkenes. Consider the electrophilic addition of H-Br to but-2-ene: The alkene abstracts a proton from the HBr, and a carbocation and bromide ion are generated. The bromide ion quickly attacks the cationic center and yields the final product. In the final product, H-Br has been added across the double bond. Ch08 Reacns of Alkenes (landscape) Page 2 Orientation of Addition Consider the addition of H-Br to 2-methylbut-2-ene: There are two possible products arising from the two different ways of adding H-Br across the double bond. But only one is observed. The observed product is the one resulting from the more stable carbocation intermediate. Tertiary carbocations are more stable than secondary. -

IB Chemistry Guide
Chemistry guide First assessment 2016 Chemistry guide First assessment 2016 Diploma Programme Chemistry guide Published February 2014 Updated February 2015 Published on behalf of the International Baccalaureate Organization, a not-for-profit educational foundation of 15 Route des Morillons, 1218 Le Grand-Saconnex, Geneva, Switzerland by the International Baccalaureate Organization (UK) Ltd Peterson House, Malthouse Avenue, Cardiff Gate Cardiff, Wales CF23 8GL United Kingdom Website: www.ibo.org © International Baccalaureate Organization 2014 The International Baccalaureate Organization (known as the IB) offers four high-quality and challenging educational programmes for a worldwide community of schools, aiming to create a better, more peaceful world. This publication is one of a range of materials produced to support these programmes. The IB may use a variety of sources in its work and checks information to verify accuracy and authenticity, particularly when using community-based knowledge sources such as Wikipedia. The IB respects the principles of intellectual property and makes strenuous efforts to identify and obtain permission before publication from rights holders of all copyright material used. The IB is grateful for permissions received for material used in this publication and will be pleased to correct any errors or omissions at the earliest opportunity. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without the prior written permission of the IB, or as expressly permitted by law or by the IB’s own rules and policy. See http://www.ibo.org/copyright. IB merchandise and publications can be purchased through the IB store at http://store.ibo.org. -

Chapter 8 - Alkenes and Alkynes II
Andrew Rosen Chapter 8 - Alkenes and Alkynes II 8.1 - Addition Reactions of Alkenes - Electrons in the π bond of alkenes react with electrophiles 8.2 - Electrophilic Addition of Hydrogen Halides to Alkenes: Mechanism and Markovnikov's Rule - Hydrogen halides can add to the double bond of alkenes - The order of reactivity of the hydrogen halides in alkene addition is HI > HBr > HCl > HF - Markovnikov's rule states that in the addition of HX to an alkene, the halide atom adds to the carbon atom of the original double bond with the fewer amount of hydrogen atoms. The hydrogen of HX will bond to the carbon atom of the greater amount of hydrogen atoms. This is precisely the same thing as stabilizing a carbocation intermediate by having the nucleophile go to the more substituted carbon - More accurately, the negative portion of an unsymmetrical ionic addition reagent will bond to the carbon of the double bond with the fewer amount of hydrogen atoms while the more positive reagent will restore the stability of the carbocation - Note that this is not in water. Typically, this will happen when the solvent is not listed - A reaction is regioselective if it can potentially yield two or more constitutional isomers but produces a predominance of one 8.3 - Stereochemistry of the Ionic Addition to an Alkene - The enantiomers are produced in equal amounts because the carbocation is trigonal planar 8.4 - Addition of Sulfuric Acid to Alkenes - Adds a hydroxide group onto the alkene via Markovnikov's Rule Allyl and Benzyl Carbocations - Resonance can stabilize a product - Revised Carbocation Stability Trend: Methyl < Primary < Secondary ≈ Primary allylic ≈ Primary benzylic < Tertiary ≈ Secondary allylic ≈ Secondary benzylic < Tertiary allylic ≈ Tertiary benzylic 1 8.5 - Addition of Water to Alkenes: Acid-Catalyzed Hydration - Note how this, too, follows Markovnikov's Rule - in is the same as + because will be a strong acid (except for ), and the solvent is in HX H2O H3O HX HF larger quantities. -

CHAPTER 3 STEREOCHEMISTRY 3.1 Geometrical Isomers in Alkenes
CHAPTER 3 STEREOCHEMISTRY Many properties of organic compounds are associated with the shape of the molecule. The "two" compounds below are isomers of Carvone, with different orientations of the isopropenyl function. One isomer (the S isomer) has the smell of spearmint whereas the other isomer (the R isomer) has the smell of caraway. The smells are a result of the way the isomers interact with receptors to send signals to the brain. Many reactions, both chemical and biological, show effects of molecular shape. O O CH3 CH3 H H CH3 CH3 R-Carvone S-Carvone Spearmint Caraway 3.1 Geometrical Isomers in Alkenes The p-bond in an alkene does not permit rotation, thus all of the atoms attached directly to the alkene lie in a plane. Groups attached to the alkene could be positioned on the same side of the alkene or on opposite sides of the alkene. Such compounds are different in chemical and physical properties as well as in their geometry, and are called geometrical isomers. In 2-butene the methyl groups can be located on the same side or on the opposite side of the double bond, giving rise to two geometrical isomers. The isomer with the methyl groups on the same side is called the cis isomer, while the isomer with the groups located on opposite sides is called the trans isomer. Trans isomers of compounds are usually more stable than cis isomers. CH CH3 CH3 3 H C C C C H H H CH3 cis- 2-butene trans- 2-butene 42 Ch 3 Stereochemistry Except for very simple alkenes with hydrogen atoms on each carbon of the alkene, the designations of cis and trans for alkenes are replaced by a system that uses E and Z designations. -
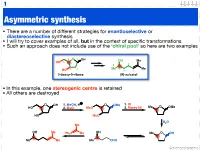
Asymmetric Synthesis
1 Asymmetric synthesis • There are a number of different strategies for enantioselective or diastereoselective synthesis • I will try to cover examples of all, but in the context of specific transformations • Such an approach does not include use of the ‘chiral pool’ so here are two examples 4 O 1 OH OH Me HO 5 5 2 3 2 Me 4 Me HO 3 1 2-deoxy-D-ribose (R)-sulcatol • In this example, one stereogenic centre is retained • All others are destroyed O OH 1. MeOH, H O OMe 1. KI HO 2. MsCl MsO 2. Raney Ni Me O OMe HO MsO H2O Me OH Me OH Ph3P Me Me O OH Me Me Me CHO Advanced organic 2 ‘Chiral pool’ II Me Me remove OH Me Me stereogenic 3 steps O O centre HO 2 4 OH PCC 3 O OH O O 6 OH HO 1 O 5 OTBDPS OTBDPS D-mannose BnO O BnO O addition of 1. NaBH protecting groups overall retention of 4 stereochemistry 2. Tf2O stereoselective Me 1. TBAF Me Me reduction Me 2. PCC Me Me O O O 3. Ph P=CHCHO NaN O 3 O 3 O N3 N3 OTf OTBDPS OTBDPS BnO O CHO BnO O BnO O Pd / C hydrogenolysis of benzyl (Bn) group & H reductive amination of resultant 2 aldehyde Me HO two step reversal of Me O 2 1 stereogenic centre reduction 1. H2, Pd / C, H of alkene & H H 3 O N 2. TFAA azide HO 4 N followed by reductive H 6 amination BnO O 5 H HO swainsonine • In this example three stereogenic centres are retained • One stereogenic centre undergoes multiple inversion -- but overall it is retained Advanced organic 3 Stereoselective reactions of alkenes • Alkenes are versatile functional groups that, as we shall see, present plenty of scope for the introduction of stereochemistry • Hydroboration permits the selective introduction of boron (surprise), which itself can undergo a wide-range of stereospecific reactions Substrate control Me H H Me H H Me 1.