LILRA2/ILT1/LIR-7) Domain Swapped Dimer: Molecular Basis for Its Non-Binding to MHC Complexes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Differential Patterns of Allelic Loss in Estrogen Receptor-Positive Infiltrating Lobular and Ductal Breast Cancer
GENES, CHROMOSOMES & CANCER 47:1049–1066 (2008) Differential Patterns of Allelic Loss in Estrogen Receptor-Positive Infiltrating Lobular and Ductal Breast Cancer L. W. M. Loo,1 C. Ton,1,2 Y.-W. Wang,2 D. I. Grove,2 H. Bouzek,1 N. Vartanian,1 M.-G. Lin,1 X. Yuan,1 T. L. Lawton,3 J. R. Daling,2 K. E. Malone,2 C. I. Li,2 L. Hsu,2 and P.L. Porter1,2,3* 1Division of Human Biology,Fred Hutchinson Cancer Research Center,Seattle,WA 2Division of Public Health Sciences,Fred Hutchinson Cancer Research Center,Seattle,WA 3Departmentof Pathology,Universityof Washington,Seattle,WA The two main histological types of infiltrating breast cancer, lobular (ILC) and the more common ductal (IDC) carcinoma are morphologically and clinically distinct. To assess the molecular alterations associated with these breast cancer subtypes, we conducted a whole-genome study of 166 archival estrogen receptor (ER)-positive tumors (89 IDC and 77 ILC) using the Affy- metrix GeneChip® Mapping 10K Array to identify sites of loss of heterozygosity (LOH) that either distinguished, or were shared by, the two phenotypes. We found single nucleotide polymorphisms (SNPs) of high-frequency LOH (>50%) common to both ILC and IDC tumors predominately in 11q, 16q, and 17p. Overall, IDC had a slightly higher frequency of LOH events across the genome than ILC (fractional allelic loss 5 0.186 and 0.156). By comparing the average frequency of LOH by chro- mosomal arm, we found IDC tumors with significantly (P < 0.05) higher frequency of LOH on 3p, 5q, 8p, 9p, 20p, and 20q than ILC tumors. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Binding Mode of the Side-By-Side Two-Igv Molecule CD226/DNAM-1 to Its Ligand CD155/Necl-5
Binding mode of the side-by-side two-IgV molecule CD226/DNAM-1 to its ligand CD155/Necl-5 Han Wanga, Jianxun Qib, Shuijun Zhangb,1, Yan Lib, Shuguang Tanb,2, and George F. Gaoa,b,2 aResearch Network of Immunity and Health (RNIH), Beijing Institutes of Life Science, Chinese Academy of Sciences (CAS), 100101 Beijing, China; and bCAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, 100101 Beijing, China Edited by K. Christopher Garcia, Stanford University School of Medicine, Stanford, CA, and approved December 3, 2018 (received for review September 11, 2018) Natural killer (NK) cells are important component of innate immu- CD226, also known as DNAM-1, belongs to the Ig superfamily nity and also contribute to activating and reshaping the adaptive and contains two extracellular Ig-like domains (CD226-D1 and immune responses. The functions of NK cells are modulated by CD226-D2), and is widely expressed in monocytes, platelets, multiple inhibitory and stimulatory receptors. Among these recep- T cells, and most of the resting NK cells (8, 13, 19, 20). The tors, the activating receptor CD226 (DNAM-1) mediates NK cell intracellular domain of CD226 does not contain a tyrosine-based activation via binding to its nectin-like (Necl) family ligand, CD155 activation motif, which is accepted as responsible for activating (Necl-5). Here, we present a unique side-by-side arrangement signal transduction of stimulatory molecules (13). Instead, it pattern of two tandem immunoglobulin V-set (IgV) domains transmits the downstream signaling by phosphorylation of in- deriving from the ectodomains of both human CD226 (hCD226- tracellular phosphorylation sites and subsequent association with ecto) and mouse CD226 (mCD226-ecto), which is substantially integrin lymphocyte function-associated antigen 1 (21). -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -

Technical Note, Appendix: an Analysis of Blood Processing Methods to Prepare Samples for Genechip® Expression Profiling (Pdf, 1
Appendix 1: Signature genes for different blood cell types. Blood Cell Type Source Probe Set Description Symbol Blood Cell Type Source Probe Set Description Symbol Fraction ID Fraction ID Mono- Lympho- GSK 203547_at CD4 antigen (p55) CD4 Whitney et al. 209813_x_at T cell receptor TRG nuclear cytes gamma locus cells Whitney et al. 209995_s_at T-cell leukemia/ TCL1A Whitney et al. 203104_at colony stimulating CSF1R lymphoma 1A factor 1 receptor, Whitney et al. 210164_at granzyme B GZMB formerly McDonough (granzyme 2, feline sarcoma viral cytotoxic T-lymphocyte- (v-fms) oncogene associated serine homolog esterase 1) Whitney et al. 203290_at major histocompatibility HLA-DQA1 Whitney et al. 210321_at similar to granzyme B CTLA1 complex, class II, (granzyme 2, cytotoxic DQ alpha 1 T-lymphocyte-associated Whitney et al. 203413_at NEL-like 2 (chicken) NELL2 serine esterase 1) Whitney et al. 203828_s_at natural killer cell NK4 (H. sapiens) transcript 4 Whitney et al. 212827_at immunoglobulin heavy IGHM Whitney et al. 203932_at major histocompatibility HLA-DMB constant mu complex, class II, Whitney et al. 212998_x_at major histocompatibility HLA-DQB1 DM beta complex, class II, Whitney et al. 204655_at chemokine (C-C motif) CCL5 DQ beta 1 ligand 5 Whitney et al. 212999_x_at major histocompatibility HLA-DQB Whitney et al. 204661_at CDW52 antigen CDW52 complex, class II, (CAMPATH-1 antigen) DQ beta 1 Whitney et al. 205049_s_at CD79A antigen CD79A Whitney et al. 213193_x_at T cell receptor beta locus TRB (immunoglobulin- Whitney et al. 213425_at Homo sapiens cDNA associated alpha) FLJ11441 fis, clone Whitney et al. 205291_at interleukin 2 receptor, IL2RB HEMBA1001323, beta mRNA sequence Whitney et al. -

Snipa Snpcard
SNiPAcard Block annotations Block info genomic range chr19:55,117,749-55,168,602 block size 50,854 bp variant count 74 variants Basic features Conservation/deleteriousness Linked genes μ = -0.557 [-4.065 – AC009892.10 , AC009892.5 , AC009892.9 , AC245036.1 , AC245036.2 , phyloP 2.368] gene(s) hit or close-by AC245036.3 , AC245036.4 , AC245036.5 , AC245036.6 , LILRA1 , LILRB1 , LILRB4 , MIR8061 , VN1R105P phastCons μ = 0.059 [0 – 0.633] eQTL gene(s) CTB-83J4.2 , LILRA1 , LILRA2 , LILRB2 , LILRB5 , LILRP1 μ = -0.651 [-4.69 – 2.07] AC008984.5 , AC008984.5 , AC008984.6 , AC008984.6 , AC008984.7 , AC008984.7 , AC009892.10 , AC009892.10 , AC009892.2 , AC009892.2 , AC009892.5 , AC010518.3 , AC010518.3 , AC011515.2 , AC011515.2 , AC012314.19 , AC012314.19 , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Mass Cytometry Analysis Reveals Complex Cell-State Modifications Of
Mass Cytometry Analysis Reveals Complex Cell-State Modifications of Blood Myeloid Cells During HIV Infection Sixtine Coindre, Nicolas Tchitchek, Lamine Alaoui, Bruno Vaslin, Christine Bourgeois, Cécile Goujard, Camille Lécuroux, Pierre Bruhns, Roger Le Grand, Anne-Sophie Beignon, et al. To cite this version: Sixtine Coindre, Nicolas Tchitchek, Lamine Alaoui, Bruno Vaslin, Christine Bourgeois, et al.. Mass Cytometry Analysis Reveals Complex Cell-State Modifications of Blood Myeloid Cells During HIV Infection. Frontiers in Immunology, Frontiers, 2019, 10.3389/fimmu.2019.02677. hal-02386129 HAL Id: hal-02386129 https://hal.archives-ouvertes.fr/hal-02386129 Submitted on 29 Nov 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License ORIGINAL RESEARCH published: 22 November 2019 doi: 10.3389/fimmu.2019.02677 Mass Cytometry Analysis Reveals Complex Cell-State Modifications of Blood Myeloid Cells During HIV Infection Sixtine Coindre 1, Nicolas Tchitchek 1, Lamine Alaoui 1, Bruno Vaslin -

(Lilrs) on Human Neutrophils: Modulators of Infection and Immunity
MINI REVIEW published: 13 May 2020 doi: 10.3389/fimmu.2020.00857 Leukocyte Immunoglobulin-Like Receptors (LILRs) on Human Neutrophils: Modulators of Infection and Immunity Alexander L. Lewis Marffy and Alex J. McCarthy* MRC Centre for Molecular Bacteriology and Infection, Imperial College London, London, United Kingdom Neutrophils have a crucial role in defense against microbes. Immune receptors allow neutrophils to sense their environment, with many receptors functioning to recognize signs of infection and to promote antimicrobial effector functions. However, the neutrophil Edited by: Nicole Thielens, response must be tightly regulated to prevent excessive inflammation and tissue damage, UMR5075 Institut de Biologie and regulation is achieved by expression of inhibitory receptors that can raise activation Structurale (IBS), France thresholds. The leukocyte immunoglobulin-like receptor (LILR) family contain activating Reviewed by: and inhibitory members that can up- or down-regulate immune cell activity. New ligands Debby Burshtyn, University of Alberta, Canada and functions for LILR continue to emerge. Understanding the role of LILR in neutrophil Tamás Laskay, biology is of general interest as they can activate and suppress antimicrobial responses University of Lübeck, Germany of neutrophils and because several human pathogens exploit these receptors for immune *Correspondence: Alex J. McCarthy evasion. This review focuses on the role of LILR in neutrophil biology. We focus on the [email protected] current knowledge of LILR expression -
Type of the Paper (Article
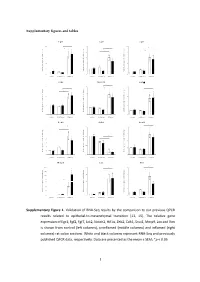
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1.