Tetrahedron, 65, 6746-6753
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UV-Curable Coating Composition
Europaisches Patentamt European Patent Office (fi) Publication number: 0 001 466 A1 Office europeen des brevets EUROPEAN PATENT APPLICATION 2y Application number: 78200215.8 © int. ci.*: C 09 D 3/49, C 08 F 299/00, C 08 F 2/50 §) Date of filing: 28.09.78 © Priority: 11.10.77 NL 7711121 Applicant: Akzo N.V., Ijssellaan 82, NL-6800 LS Arnhem (NL) (7g) Inventor: Noomen, Arie, Schoonoord 22, NL-2215 Ed @ Date of publication of application: 18.04.79 Voorhout (NL) Bulletin 79/8 Inventor: Wolters, Egbert, Potgieterstraat 21, NL-1053 XP Amsterdam (NL) @ Representative: Sieders, Rene et ai, P.O.Box 314, @ Designated Contracting States: BE DE FR GB NL SE NL-6800 AH Arnhem (NL) @ U.V.-curable coating composition. The invention relates to a coating composition which is curable under the influence of ultraviolet light and which comprises a U.V.-curable binder, a photoinitiator and as accelerator a tetrahydro-1,3-oxazine compound and/or an oxazolidine compound. The U.V.-curable binder is pref- erably an adduct containing at least one isocyanate group of a) an acrylic or methacrylic hydroxy ester having 5 to 20 carbon atoms and b) a polyisocyanate having 4 to 40 carbon atoms and 2 to 4 isocyanate groups. The invention relates to a coating composition which can be cured under the influence of ultraviolet light ;and is based on a U.V.-curable binder, a photoinitiator and a nitrogen-containing accelerator. In known com- positions useismade generally of an aromatic carbonyl compound as photoinitiator, and of an alkanolamine as accelerator. -

Phamarcological Effects Produced by the Modifications in Isoxazole Moeities
Archana c. et al. / Asian Journal of Phytomedicine and Clinical Research. 4(1), 2016, 29 - 32. Review Article CODEN: AJPCFF ISSN: 2321 – 0915 Asian Journal of Phytomedicine and Clinical Research Journal home page: www.ajpcrjournal.com PHAMARCOLOGICAL EFFECTS PRODUCED BY THE MODIFICATIONS IN ISOXAZOLE MOEITIES C. Archana *1 , E. Akila 2, Priya john 3 1*Department of Pharmaceutical Chemistry, Prime College of Pharmacy, Palakkad, Kerala, India. 2Department of Pharmacognosy, Prime College of Pharmacy, Palakkad, Kerala, India. 3Department of Pharmacology, Prime College of Pharmacy, Palakkad, Kerala, India. ABSTRACT Isoxazole and its derivatives are an important class of heterocyclic compound displaying a broad spectrum of biological activities which have made them privileged structures. In the present work, attempts were made to identify leading isoxazole moieties as candidate drugs against many diseases. For example, isoxazole substituted 9-anilino acridine derivatives was found to have increased antioxidant activities. The molecular docking studies show a good correlation between their biological activities screened and auto dock binding free energy. These derivatives will encourage helping to design future anti cancer agents with higher therapeutic potential. Another example is isoxazole incorporated 2-quinolones to show increased antimicrobial and anti inflammatory activities. More importantly, various isoxazole derivatives greatly increase biological properties of the structure like anti-infective action, anticancer properties, anti-protozoal and mutagenic properties. A modification in their structures has offered a high degree of diversity that has proven useful for the development of new therapeutic agents having improved potency and lesser toxicity. In the present study of concise review, is provided on the activities of isoxazole and its derivatives which involve history, chemistry, different methods of synthesis of isoxazole with biological activities and docking studies. -

Isoxazoles: Molecules with Potential Medicinal Properties
IJPCBS 2013, 3(2), 294-304 Jayaroopa et al. ISSN: 2249-9504 INTERNATIONAL JOURNAL OF PHARMACEUTICAL, CHEMICAL AND BIOLOGICAL SCIENCES Available online at www.ijpcbs.com Research Article ISOXAZOLES: MOLECULES WITH POTENTIAL MEDICINAL PROPERTIES K. Ajay Kumar and P. Jayaroopa* Post Graduate Department of Chemistry, Yuvaraja’s College, University of Mysore, Mysore-570 005, India. ABSTRACT Isoxazole is a five membered heterocyclic compound having various pharmacological actions. The great interest associated with isoxazoles and their derivatives is based on their versatility as synthetic building blocks, their latent functionalities as enaminones, 1,3-dicarbonyl compounds, γ-amino alcohols, and β-hydroxy nitriles have been widely exploited for the synthesis of other heterocycles and complex molecules. This review paper comprises of up to date information on isoxazole analogs. More emphasis was given to critical discussion on the synthetic strategy of isoxazole derivative, their utility as building blocks in their transformation to more biologically potent molecules. Results of isoxazole derivatives and their substitutions effect on diverse biological activities also presented. Keywords: Isoxazoles, antioxidant, antimicrobial, analgesic, anti-platelet, anti-HIV. INTRODUCTION sulfisoxazole, oxacillin, cycloserine and acivicin Nitrogen containing heterocycles with an have been in commercial use for many years. oxygen atom are considered as an important Cycloserine is the best known antibiotic drug class of compounds in medicinal chemistry that possess antitubercular, antibacterial because of their diversified biological activities and in treatment of leprosy. Acivicin is applications. The exploitation of a simple an antitumour, antileishmania drug, while molecule with different functionalities for the isoxaflutole is used as herbicidal drug. synthesis of heterocycles is a worthwhile Isoxazoles have illustrious history; their contribution in the chemistry of heterocycles. -

Matrix Scientific PO BOX 25067 COLUMBIA, SC 29224-5067 Telephone: 803-788-9494 Fax: 803-788-9419 SAFETY DATA SHEET Transportation Emergency: 3E Co
Matrix Scientific PO BOX 25067 COLUMBIA, SC 29224-5067 Telephone: 803-788-9494 Fax: 803-788-9419 SAFETY DATA SHEET Transportation Emergency: 3E Co. (5025) 800-451-8346 1. Product Identification Name Oxazolidine-2,5-dione Catalog Number 094908 CAS Registry Number [2185-00-4] Company Matrix Scientific Physical Address 131 Pontiac Business Center Drive Elgin, SC 29045 USA Telephone/Fax (803)788-9494/(803)788-9419 2. Hazard Identification Hazardous Ingredients Oxazolidine-2,5-dione GHS label elements, including precautionary statements Pictogram Signal word WARNING Hazard statement(s) H317 May cause an allergic skin reaction H319 Causes serious eye irritation Precautionary statement(s) P280 Wear protective gloves/protective clothing/eye protection/face protection. P305+351+338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses if present and easy to do - continue rinsing. 3. Composition, Information or Ingredients Name Oxazolidine-2,5-dione 4. First Aid Measures Eye Contact: Check for and remove any contact lenses. Immediately flush 1 Last Updated 2/11/2017 eyes with clean, running water for at least 15 minutes while keeping eyes open. Cool water may be used. Seek medical attention. Skin Contact: After contact with skin, wash with generous quantities of running water. Gently and thoroughly wash affected area with running water and non- abrasive soap. Cool water may be used. Cover the affected area with emollient. Seek medical attention. Wash any contaminated clothing prior to reusing. Inhalation: Remove the victim from the source of exposure to fresh, uncontaminated air. If victim's breathing is difficult, administer oxygen. Seek medical attention. -

A New Class of Anticancer Agents
Chem Biol Drug Des 2014; 83: 126–131 Research Letter Mefloquine–Oxazolidine Derivatives: A New Class of Anticancer Agents Felipe A. R. Rodrigues1, Igor da S. Bomfim1, applications in other fields, such as against Gram-positive Bruno C. Cavalcanti1, Claudia Pessoa1, Raoni and Gram-negative bacteria (1), in particular against Myco- S. B. Goncalves2, James L. Wardell2,3, Solange bacterium tuberculosis (2,3) and Mycobacterium avium M. S. V. Wardell4 and Marcus V. N. de Souza2,* complex (MAC) (4,5) and also against schistosomiasis (6), neurodegenerative, and (neuro-) inflammatory diseases (7). 1Laboratorio de Oncologia Experimental, Universidade A clear indication of the promise of this drug in fields other Federal do Ceara, Fortaleza, CE 3157, Brazil than malaria is the number of recent patent applications 2FioCruz-Fundacßao~ Oswaldo Cruz, Instituto de Tecnologia (5,7,8). More recently, much attention has been paid to the em Farmacos-Far-Manguinhos, Rua Sizenando Nabuco, potential perspectives of MQ as an anticancer agent. As 100, Manguinhos, 21041-250, Rio de Janeiro, RJ 35513, estimated by the National Institutes of Health (NIH), the over- Brazil all costs of cancer in 2007 were $226.8 billion.a Cancer is a 3Department of Chemistry, University of Aberdeen, Old leading cause of deaths worldwide and accounted for 7.6 Aberdeem, Aberdeem, AB 24 3UE, UK b 4CHEMSOL, 1 Harcourt Road, Aberdeen, AB15 5NY, UK million deaths (13% of all deaths) in 2008. Louie and *Corresponding author: Marcus V. N. de Souza, coworkers (9) found MQ to be the most potent anticancer marcos_souza@far.fiocruz.br drug when compared to chloroquine and the fluoroquino- lone drugs cipro and levofloxacin. -

United States Patent (19) 11 Patent Number: 5,977,285 Ernst MOOS Et Al
USOO5977285A United States Patent (19) 11 Patent Number: 5,977,285 Ernst MOOS et al. (45) Date of Patent: Nov. 2, 1999 54 SPRAYABLE COATING COMPOSITIONS 992721 5/1965 United Kingdom. WO 92/13907 8/1992 WIPO ............................ CO8G 18/10 COMPRISING OXAZOLIDINES, ISOCYANATES AND HYDROXYL OR AMINE WO 93/17060 9/1993 WIPO ............................ CO8G 63/02 FUNCTIONAL RESNS WO 94/27746 12/1994 WIPO .............................. BO5D 7/24 WO95/14528 6/1995 WIPO .............................. B01J 13/OO (75) Inventors: Jan Wilhelm Ernst Moos, Bloomfield WO 96/083O8 3/1996 WIPO .............................. B01J 13/OO Hills; Heide Anne Lochbiler, East OTHER PUBLICATIONS Pointe; Jason Donald Weaver, New Angus Chemical Company Technical Bulletin TB 91 Zol Baltimore; Latoska Nikita Price, dine(R) RD-20, 1–8 (1994). Southfield; Frances Lamb, Northville; International Search Report, dated Nov. 27, 1998 (PCT/ Donald Lynn Rutledge, Jr., Clawson, EP98/04942). all of Mich.; Ann Alfred Johanna International Search Report, dated Dec. 4, 1998 (PCT/EP98/ Lemaire, Leiderdorp, Netherlands; 04943). Antonius Hendrikus Gerardus Van International Search Report, dated Dec. 11, 1998 (PCT/ Engelen, Alphen A/D Rijn, Netherlands; EP98/04944). Catharine Marie den Breejen, Rohm and Haas Comppany Acryloid(R) Resins, Maintenance Dordrecht, Netherlands and Marine Coatings, Experimental Reactive Modifier QM-1007M, Reactive Diluent for Increasing Paint Solids Assignee: Akzo Nobel N.V., Netherlands and Improving Film Build of Acrylic-Urethane Coating, 1990, pp. 1–20. Appl. No.: 08/906,645 Donald C. Schall, High Solids Isocyanate-Oxazolidine Filed: Aug. 7, 1997 Coatings, Presented at the Water-Borne & High-Solids coatings Symposium, Feb. 13-15, 1985, New Orleans, Loui Int. -

Chemicals Subject to TSCA Section 12(B) Export Notification Requirements (January 16, 2020)
Chemicals Subject to TSCA Section 12(b) Export Notification Requirements (January 16, 2020) All of the chemical substances appearing on this list are subject to the Toxic Substances Control Act (TSCA) section 12(b) export notification requirements delineated at 40 CFR part 707, subpart D. The chemicals in the following tables are listed under three (3) sections: Substances to be reported by Notification Name; Substances to be reported by Mixture and Notification Name; and Category Tables. TSCA Regulatory Actions Triggering Section 12(b) Export Notification TSCA section 12(b) requires any person who exports or intends to export a chemical substance or mixture to notify the Environmental Protection Agency (EPA) of such exportation if any of the following actions have been taken under TSCA with respect to that chemical substance or mixture: (1) data are required under section 4 or 5(b), (2) an order has been issued under section 5, (3) a rule has been proposed or promulgated under section 5 or 6, or (4) an action is pending, or relief has been granted under section 5 or 7. Other Section 12(b) Export Notification Considerations The following additional provisions are included in the Agency's regulations implementing section 12(b) of TSCA (i.e. 40 CFR part 707, subpart D): (a) No notice of export will be required for articles, except PCB articles, unless the Agency so requires in the context of individual section 5, 6, or 7 actions. (b) Any person who exports or intends to export polychlorinated biphenyls (PCBs) or PCB articles, for any purpose other than disposal, shall notify EPA of such intent or exportation under section 12(b). -
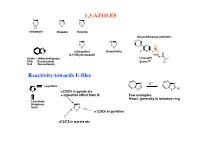
1,3-AZOLES Reactivity Towards E-Files
1,3-AZOLES N N N N S O H Imidazole Thiazole Oxazole Oxazolidineone antibiotic N HN O N O N O O 2-Oxazoline Oxazolidine F N O X (4,5-Dihydrooxazol) H N X=NH: 1H-Benzimidazole Linesolid X=O: Benzoxazole ZyvoxTM O X=S Benzotiazole Reactivity towards E-files N E+ N N ≈ pyridine X X E X ≈C3/C4 in pyrole etc + inductive effect from N Few examples React. generally in benzene ring ≈ pyrazole thiophene N furan X ≈ C2/C6 in pyridine ≈C2/C5 in pyrole etc Reaction with electrophiles on N - Protonation H H pKa H N H+ N N N H X=NH: 7.1 N X X X=S: 2.5 N N X=O: 0.8 H H O stabilized Destabilized R R N HN Taut.: N N H R= Me: ca 1 : 1 R=NO2: ca 400 : 1 Reaction with electrophiles on N R 'R R 'R R N-Alkylation N N N N N N H H R'-X R N R-X N - H+ R R R X X HN HN N May be sterically favoured Reactivity: N N N R' R' X = N-Me X=S X=O 900 : 15 : 1 R R R N N N N low react. N N N N H R' SO2Ph Base R'-X - H+ R R 'R R HN N N N N N N N H Base Et Et N + - N N PhCOCl 1) Et3O BF4 N N N O O HN N steric control Reaction with electrophiles on N N-Acylation Only rel. -

WO 2010/021920 Al
(12) INTERNATIONALAPPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 25 February 2010 (25.02.2010) WO 2010/021920 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C08L 67/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, (21) International Application Number: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, PCT/US2009/053829 DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, 14 August 2009 (14.08.2009) KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (25) Filing Language: English NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (26) Publication Language: English SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/090,283 20 August 2008 (20.08.2008) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant (for all designated States except US): RHEIN GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, CHEMIE CORPORATION [US/US]; 145 Parker ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, Court, Chardon, OH 44024 (US). -

Recent Syntheses of Steroidal Oxazoles, Oxazolines and Oxazolidines Besma Bendif, Malika Ibrahim-Ouali, Frédéric Dumur
Recent syntheses of steroidal oxazoles, oxazolines and oxazolidines Besma Bendif, Malika Ibrahim-Ouali, Frédéric Dumur To cite this version: Besma Bendif, Malika Ibrahim-Ouali, Frédéric Dumur. Recent syntheses of steroidal oxazoles, oxa- zolines and oxazolidines. ARKIVOC - Online Journal of Organic Chemistry, Arkat USA Inc, 2021, 2021 (1), pp.471-490. 10.24820/ark.5550190.p011.512. hal-03230347 HAL Id: hal-03230347 https://hal.archives-ouvertes.fr/hal-03230347 Submitted on 19 May 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. The Free Internet Journal Review for Organic Chemistry Archive for Arkivoc 2021, part i, 471-490 Organic Chemistry Recent syntheses of steroidal oxazoles, oxazolines and oxazolidines Besma Bendif,a,b Malika Ibrahim-Ouali,*a and Frédéric Dumur c aAix Marseille Univ, CNRS, Centrale Marseille, iSm2, F-13397 Marseille, France bLaboratoire de Chimie Organique Appliquée, Faculté des Sciences, Université de Guelma, Algeria cAix Marseille Univ, CNRS, ICR, UMR 7273, F-13397 Marseille, France Email address: [email protected] Received 03-15-2021 Accepted 04-11-2021 Published on line 05-08-2021 Abstract It was found that the introduction of heterocycles to steroids often leads in a change of their physiological activity and the appearance of new interesting biological precursors. -

Heterocyclic Anticancer Compounds: Recent Advances and the Paradigm Shift Towards the Use of Nanomedicine’S Tool Box
Molecules 2015, 20, 16852-16891; doi:10.3390/molecules200916852 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Heterocyclic Anticancer Compounds: Recent Advances and the Paradigm Shift towards the Use of Nanomedicine’s Tool Box Pedro Martins 1, João Jesus 1, Sofia Santos 1, Luis R. Raposo 1, Catarina Roma-Rodrigues 1, Pedro Viana Baptista 1,* and Alexandra R. Fernandes 1,2,* 1 UCIBIO, Departamento de Ciências da Vida, Faculdade de Ciências e Tecnologia da Universidade Nova de Lisboa, Campus de Caparica, 2829-516 Caparica, Portugal; E-Mails: [email protected] (P.M.); [email protected] (J.J.); [email protected] (S.S.); [email protected] (L.R.R.); [email protected] (C.R.-R.) 2 Centro de Química Estrutural, Complexo 1, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal * Authors to whom correspondence should be addressed; E-Mails: [email protected] (P.V.B.); [email protected] (A.R.F.); Tel./Fax: +351-21-2894-8530 (P.V.B. & A.R.F.). Academic Editor: Richard A. Bunce Received: 19 June 2015 / Accepted: 9 September 2015 / Published: 16 September 2015 Abstract: The majority of heterocycle compounds and typically common heterocycle fragments present in most pharmaceuticals currently marketed, alongside with their intrinsic versatility and unique physicochemical properties, have poised them as true cornerstones of medicinal chemistry. Apart from the already marketed drugs, there are many other being investigated for their promising activity against several malignancies. In particular, anticancer research has been capitalizing on the intrinsic versatility and dynamic core scaffold of these compounds. -

Research Themes
Project Title: Designing palladium based N−heterocyclic carbene complexes for anticancer application Project Number IMURA0719 Monash Main Supervisor Prof. Glen Deacon (Name, Email Id, Phone) [email protected] Full name, Email Monash Co-supervisor(s) Prof. Bayden Wood (Name, Email Id, Phone) [email protected] Monash Head of Dr David Turner (grad coordinator chemistry) Dept/Centre (Name,Email) Full name, email [email protected] Monash Department: Chemistry Monash ADRT Prof Peter Betts Full name, email (Name,Email) [email protected] IITB Main Supervisor Prof. Prasenjit Ghosh Full name, Email (Name, Email Id, Phone) [email protected] IITB Co-supervisor(s) (Name, Email Id, Phone) Full name, Email IITB Head of Dept Prof. Krishna P. Kaliappan (Name, Email, Phone) Full name, email [email protected] IITB Department: Department of Chemistry Research Clusters: Research Themes: Highlight which of the Academy’s Highlight which of the Academy’s Theme(s) this CLUSTERS this project will address? project will address? (Please nominate JUST one. For more information, see (Feel free to nominate more than one. For more information, see www.iitbmonash.org) www.iitbmonash.org) 1 Material Science/Engineering (including Nano, Metallurgy) 1 Advanced computational engineering, simulation and manufacture 2 Energy, Green Chem, Chemistry, Catalysis, Reaction Eng 2 Infrastructure Engineering 3 Math, CFD, Modelling, Manufacturing 4 CSE, IT, Optimisation, Data, Sensors, Systems, 3 Clean Energy Signal Processing, Control 5 Earth Sciences and