Plasmodium Falciparum Malaria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Membrane Complement Regulatory Protein CD59 and Its Association with Rheumatoid Arthritis and Systemic Lupus Erythematosus
Current Medicine Research and Practice 9 (2019) 182e188 Contents lists available at ScienceDirect Current Medicine Research and Practice journal homepage: www.elsevier.com/locate/cmrp Review Article The membrane complement regulatory protein CD59 and its association with rheumatoid arthritis and systemic lupus erythematosus * Nibhriti Das a, Devyani Anand a, Bintili Biswas b, Deepa Kumari c, Monika Gandhi c, a Department of Biochemistry, All India Institute of Medical Sciences, New Delhi 110029, India b Department of Zoology, Ramjas College, University of Delhi, India c University School of Biotechnology, Guru Gobind Singh Indraprastha University, India article info abstract Article history: The complement cascade consisting of about 50 soluble and cell surface proteins is activated in auto- Received 8 May 2019 immune inflammatory disorders. This contributes to the pathological manifestations in these diseases. In Accepted 30 July 2019 normal health, the soluble and membrane complement regulatory proteins protect the host against Available online 5 August 2019 complement-mediated self-tissue injury by controlling the extent of complement activation within the desired limits for the host's benefit. CD59 is a membrane complement regulatory protein that inhibits the Keywords: formation of the terminal complement complex or membrane attack complex (C5b6789n) which is CD59 generated on complement activation by any of the three pathways, namely, the classical, alternative, and RA SLE the mannose-binding lectin pathway. Animal experiments and human studies have suggested impor- Pathophysiology tance of membrane complement proteins including CD59 in the pathophysiology of rheumatoid arthritis Disease marker (RA) and systemic lupus erythematosus (SLE). Here is a brief review on CD59 and its distribution, structure, functions, and association with RA and SLE starting with a brief introduction on the com- plement system, its activation, the biological functions, and relations of membrane complement regu- latory proteins, especially CD59, with RA and SLE. -

An Anticomplement Agent That Homes to the Damaged Brain and Promotes Recovery After Traumatic Brain Injury in Mice
An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice Marieta M. Rusevaa,1,2, Valeria Ramagliab,1, B. Paul Morgana, and Claire L. Harrisa,3 aInstitute of Infection and Immunity, School of Medicine, Cardiff University, Cardiff CF14 4XN, United Kingdom; and bDepartment of Genome Analysis, Academic Medical Center, Amsterdam 1105 AZ, The Netherlands Edited by Douglas T. Fearon, Cornell University, Cambridge, United Kingdom, and approved September 29, 2015 (received for review July 15, 2015) Activation of complement is a key determinant of neuropathology to rapidly and specifically inhibit MAC at sites of complement and disability after traumatic brain injury (TBI), and inhibition is activation, and test its therapeutic potential in experimental TBI. neuroprotective. However, systemic complement is essential to The construct, termed CD59-2a-CRIg, comprises CD59a linked fight infections, a critical complication of TBI. We describe a to CRIg via the murine IgG2a hinge. CD59a prevents assembly targeted complement inhibitor, comprising complement receptor of MAC in cell membranes (16), whereas CRIg binds C3b/iC3b of the Ig superfamily (CRIg) fused with complement regulator CD59a, deposited at sites of complement activation (17). The IgG2a designed to inhibit membrane attack complex (MAC) assembly at hinge promotes dimerization to increase ligand avidity. CD59- sites of C3b/iC3b deposition. CRIg and CD59a were linked via the 2a-CRIg protected in the TBI model, demonstrating that site- IgG2a hinge, yielding CD59-2a-CRIg dimer with increased iC3b/C3b targeted anti-MAC therapeutics may be effective in prevention binding avidity and MAC inhibitory activity. CD59-2a-CRIg inhibited of secondary neuropathology and improve neurologic recovery MAC formation and prevented complement-mediated lysis in vitro. -

Complement Herbert L
Host Defense 2011 Complement Herbert L. Mathews, Ph.D. COMPLEMENT Date: 4/11/11 Reading Assignment: Janeway’s Immunobiology, 7th Edition, pp. 54-55, 61-82, 406- 409, 514-515. Figures: (Unless otherwise noted) Janeway’s Immunobiology, 7th Edition, Murphy et al., Garland Publishing. KEY CONCEPTS AND LEARNING OBJECTIVES You will be able to describe the mechanism and consequences of the activation of the complement system. To attain the goals for these lectures you will be able to: a. List the components of the complement system. b. Describe the three activation pathways for complement. c. Explain the consequences of complement activation. d. Describe the consequence of complement deficiency. Page 1 Host Defense 2011 Complement Herbert L. Mathews, Ph.D. CONTENT SUMMARY Introduction Nomenclature Activation of Complement The classical pathway The mannan-binding lectin pathway The alternative pathway Biological Consequence of Complement Activation Cell lysis and viral neutralization Opsonization Clearance of Immune Complexes Inflammation Regulation of Complement Activation Human Complement Component Deficiencies Page 2 Host Defense 2011 Complement Herbert L. Mathews, Ph.D. Introduction The complement system is a group of more than 30 plasma and membrane proteins that play a critical role in host defense. When activated, complement components interact in a highly regulated fashion to generate products that: Recruit inflammatory cells (promoting inflammation). Opsonize microbial pathogens and immune complexes (facilitating antigen clearance). Kill microbial pathogens (via a lytic mechanism known as the membrane attack complex). Generate an inflammatory response. Complement activation takes place on antigenic surfaces. However, the activation of complement generates several soluble fragments that have important biologic activity. -

Targeting of Mannan-Binding Lectin-Associated Serine Protease-2 Confers Protection from Myocardial and Gastrointestinal Ischemia/Reperfusion Injury
Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury Wilhelm J. Schwaeblea,1, Nicholas J. Lyncha, James E. Clarkb, Michael Marberb, Nilesh J. Samanic, Youssif Mohammed Alia,d, Thomas Dudlere, Brian Parente, Karl Lhottaf, Russell Wallisa, Conrad A. Farrarg, Steven Sacksg, Haekyung Leeh, Ming Zhangh, Daisuke Iwakii, Minoru Takahashii, Teizo Fujitai, Clark E. Tedforde, and Cordula M. Stovera Departments of aInfection, Immunity, and Inflammation and cCardiovascular Sciences, University of Leicester, Leicester LE1 9HN, United Kingdom; bBritish Heart Foundation Centre and gMedical Research Council Centre for Transplantation and National Institute for Health Research Biomedical Research Centre at Guy’s and St. Thomas’ National Health Service Foundation Trust, King’s College London, London SE1 9RT, United Kingdom; dFaculty of Pharmacy, Department of Microbiology, University of Mansoura, Mansoura 35516, Egypt; eOmeros Corporation, Seattle, WA 98101; fLandeskrankenhaus Feldkirch, 6807 Feldkirch, Austria; hDepartment of Anesthesiology, State University of New York-Downstate Medical Center, New York, NY 11203; and iDepartment of Immunology, Fukushima Medical University, Fukushima 960-1295, Japan Edited* by Douglas T. Fearon, University of Cambridge School of Clinical Medicine, Cambridge, United Kingdom, and approved March 16, 2011 (received for review February 1, 2011) Complement research experienced a renaissance with the discovery aberrant glycosylation -
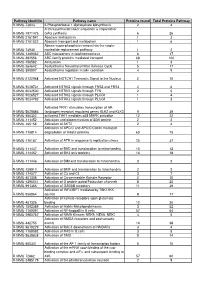
RAW PIPPR Signature Pathway List
Pathway identifier Pathway name Proteins found Total Proteins Pathway R-MMU-73843 5-Phosphoribose 1-diphosphate biosynthesis 2 4 A tetrasaccharide linker sequence is required for R-MMU-1971475 GAG synthesis 6 26 R-MMU-2161541 Abacavir metabolism 1 3 R-MMU-2161522 Abacavir transport and metabolism 1 6 Abasic sugar-phosphate removal via the single- R-MMU-73930 nucleotide replacement pathway 1 2 R-MMU-1369062 ABC transporters in lipid homeostasis 6 17 R-MMU-382556 ABC-family proteins mediated transport 68 100 R-MMU-156582 Acetylation 1 3 R-MMU-264642 Acetylcholine Neurotransmitter Release Cycle 5 17 R-MMU-399997 Acetylcholine regulates insulin secretion 4 9 R-MMU-2122948 Activated NOTCH1 Transmits Signal to the Nucleus 2 10 R-MMU-9028731 Activated NTRK2 signals through FRS2 and FRS3 2 6 R-MMU-9032500 Activated NTRK2 signals through FYN 2 5 R-MMU-9026527 Activated NTRK2 signals through PLCG1 1 4 R-MMU-9034793 Activated NTRK3 signals through PLCG1 1 3 Activated PKN1 stimulates transcription of AR R-MMU-5625886 (androgen receptor) regulated genes KLK2 and KLK3 9 39 R-MMU-450302 activated TAK1 mediates p38 MAPK activation 12 22 R-MMU-111452 Activation and oligomerization of BAK protein 2 2 R-MMU-165158 Activation of AKT2 2 4 Activation of APC/C and APC/C:Cdc20 mediated R-MMU-176814 degradation of mitotic proteins 63 75 R-MMU-176187 Activation of ATR in response to replication stress 25 37 R-MMU-111447 Activation of BAD and translocation to mitochondria 10 12 R-MMU-114452 Activation of BH3-only proteins 14 18 R-MMU-111446 Activation of BIM and -

Current Understanding of the Role of Complement in Iga Nephropathy
BRIEF REVIEW www.jasn.org Current Understanding of the Role of Complement in IgA Nephropathy † ‡ Nicolas Maillard,* Robert J. Wyatt, Bruce A. Julian,* Krzysztof Kiryluk,§ Ali Gharavi,§ | Veronique Fremeaux-Bacchi, and Jan Novak* *University of Alabama at Birmingham, Departments of Microbiology and Medicine, Birmingham, Alabama; †Université Jean Monnet, Groupe sur l9immunité des Muqueuses et Agents Pathogènes, St. Etienne, Pôle de Recherche et d9Enseignement Supérieur, Université de Lyon, Lyon, France; ‡University of Tennessee Health Science Center and Children’s Foundation Research at the Le Bonheur Children’s Hospital, Memphis, Tennessee; §Columbia University, Department of Medicine, New York, New York; and |Unité Mixte de Recherche en Santé 1138, Team “Complement and Diseases,” Cordeliers Research Center, Paris, France ABSTRACT Complement activation has a role in the pathogenesis of IgA nephropathy, an IgA1 autoantibodies targeting terminal autoimmune disease mediated by pathogenic immune complexes consisting of N-acetylgalactosamine in the hinge region galactose-deficient IgA1 bound by antiglycan antibodies. Of three complement- of Gd-IgA1.8 The third hit is the formation activation pathways, the alternative and lectin pathways are involved in IgA of Gd-IgA1–containing circulating im- nephropathy. IgA1 can activate both pathways in vitro, and pathway components mune complexes, some of which may de- are present in the mesangial immunodeposits, including properdin and factor H in the positintheglomeruliandinciteinjury(hit alternative pathway and mannan-binding lectin, mannan–binding lectin–associated four), potentially leading to chronic kidney serine proteases 1 and 2, and C4d in the lectin pathway. Genome–wide association damage. Other proteins, such as the solu- studies identified deletion of complement factor H–related genes 1 and 3 as protective ble form of Fca receptor, can bind against the disease. -

SARS-Cov-2: Pathogenesis, and Advancements in Diagnostics and Treatment
REVIEW published: 06 October 2020 doi: 10.3389/fimmu.2020.570927 SARS-CoV-2: Pathogenesis, and Advancements in Diagnostics and Treatment Khalil Khalaf 1*, Natalia Papp 1, Jadzia Tin-Tsen Chou 1, Doris Hana 1, Andrzej Mackiewicz 1,2 and Mariusz Kaczmarek 1,2 1 Department of Cancer Immunology, Poznan University of Medical Sciences, Poznan,´ Poland, 2 Department of Cancer Diagnostics and Immunology, Greater Poland Cancer Center, Poznan,´ Poland The emergence and rapid spread of SARS-CoV-2 in December 2019 has brought the world to a standstill. While less pathogenic than the 2002–2003 SARS-CoV, this novel betacoronavirus presents a global threat due to its high transmission rate, ability to invade multiple tissues, and ability to trigger immunological hyperactivation. The identification of the animal reservoir and intermediate host were important steps toward slowing the spread of disease, and its genetic similarity to SARS-CoV has helped to determine pathogenesis and direct treatment strategies. The exponential increase in cases has Edited by: necessitated fast and reliable testing procedures. Although RT-PCR remains the gold Xin-Xin Zhang, standard, it is a time-consuming procedure, paving the way for newer techniques such Shanghai Public Health Clinical as serologic tests and enzyme immunoassays. Various clinical trials using broad antiviral Center, China Reviewed by: agents in addition to novel medications have produced controversial results; however, Kartika Padhan, the advancement of immunotherapy, particularly monoclonal antibodies and immune National Institutes of Health (NIH), modulators is showing great promise in clinical trials. Non-orthodox medications such as United States Youhua Xie, anti-malarials have been tested in multiple institutions but definitive conclusions are yet Shanghai Medical College of Fudan to be made. -

Complement Regulator CD46: Genetic Variants and Disease Associations M
Liszewski and Atkinson Human Genomics (2015) 9:7 DOI 10.1186/s40246-015-0029-z REVIEW Open Access Complement regulator CD46: genetic variants and disease associations M. Kathryn Liszewski* and John P. Atkinson Abstract Membrane cofactor protein (MCP; CD46) is an ubiquitously expressed complement regulatory protein that protects host cells from injury by complement. This type-I membrane glycoprotein serves as a cofactor for the serine protease factor I to mediate inactivation of C3b and C4b deposited on host cells. More than 60 disease-associated mutations in MCP have now been identified. The majority of the mutations are linked to a rare thrombotic microangiopathic-based disease, atypical hemolytic uremic syndrome (aHUS), but new putative links to systemic lupus erythematosus, glomerulonephritis, and pregnancy-related disorders among others have also been identified. This review summarizes our current knowledge of disease-associated mutations in this complement inhibitor. Keywords: CD46, Membrane cofactor protein, Complement, Complement regulation, Atypical hemolytic uremic syndrome Introduction than five minutes. The most important function of these The complement system is one of the most ancient com- covalently deposited complement proteins is to opsonize ponents of innate immunity. It likely evolved from a C3- the target, thus serving as ligands for complement recep- like protein that was cleaved by proteases into biologically tors on peripheral blood cells; specifically, erythrocytes, active self-defense fragments to counteract invading mi- neutrophils, B lymphocytes and monocytes, as well as crobes, particularly bacteria, and to clear biologic debris dendritic cells, and tissue monocytes. The receptors serve (self and foreign) [1, 2]. The development of a circulatory to bind (immune adherence), internalize (in some cases), system may have been the key evolutionary pressure that transport, and clear the microbe. -

Activity of Mannose-Binding Lectin on Bacterial-Infected Chickens—A Review
animals Review Activity of Mannose-Binding Lectin on Bacterial-Infected Chickens—A Review Peter A. Idowu 1,* , Adeola P. Idowu 2, Oliver T. Zishiri 3 , Takalani J. Mpofu 1 , Edwin J. A. Veldhuizen 4 , Khathutshelo A. Nephawe 1 and Bohani Mtileni 1 1 Department of Animal Sciences, Tshwane University of Technology, Private Bag X680, Pretoria 0001, South Africa; [email protected] (T.J.M.); [email protected] (K.A.N.); [email protected] (B.M.) 2 Department of Animal Science, North West University, Private Bag X2046, Mmabatho 2735, South Africa; [email protected] 3 Discipline of Genetics, School of Life Sciences, University of KwaZulu-Natal, Private Bag X54001, Durban 4000, South Africa; [email protected] 4 Department of Biomolecular Health Sciences, Division Infectious Diseases and Immunology, Section of Immunology, Faculty of Veterinary Medicine, Utrecht University, 3584 CS Utrecht, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +27-71-042-3992 Simple Summary: In the quest to combat bacterial-related diseases in chickens, different methods, of which some are less economical and less effective on the long-term, have been adapted. However, chickens possess mannose-binding lectin (MBL) which could be vital in managing pathogenic bacteria in chickens. MBL is one of the soluble proteins secreted by the chicken’s innate immune system which can be activated when chickens are exposed to chicken-related diseases. This review explains Citation: Idowu, P.A.; Idowu, A.P.; how mannose-binding lectin activation can help in fighting bacterial pathogens in chickens. This Zishiri, O.T.; Mpofu, T.J.; Veldhuizen, E.J.A.; Nephawe, K.A.; Mtileni, B. -

Lectin Pathway of Complement Activation and Relation with Clinical Complications in Critically Ill Children
nature publishing group Clinical Investigation Articles Lectin pathway of complement activation and relation with clinical complications in critically ill children Catherine Ingels1, Ilse Vanhorebeek1, Rudi Steffensen2, Inge Derese1, Lisbeth Jensen3, Pieter J. Wouters1, Greet Hermans1, Steffen Thiel3 and Greet Van den Berghe1 BACKGROUND: Critically ill children are susceptible to nos- variants of innate immunity genes would thus orchestrate dif- ocomial infections, which contribute to adverse outcomes. ferent responses to apparently similar insults (3,4). Deficiencies in the innate immunity lectin pathway of The innate immune system represents the immediate response complement activation are implicated in a child’s vulner- toward injury, before amplification of the adaptive immune sys- ability to infections in conditions such as cancer, but the role tem. Part of innate immunity is started when membrane-bound, during critical illness remains unclear. We hypothesized that soluble, or intracellular receptor proteins recognize highly con- low on-admission levels of the pathway proteins are, in part, served pathogen- or danger-associated molecular patterns genetically determined and associated with susceptibility to (5). Mannose-binding lectin (MBL), H-ficolin, and M-ficolin infectious complications and adverse outcomes. are examples of soluble pattern recognition molecules, able to METHODS: We studied protein levels of mannose-binding bind certain patterns of chemical structures on microorgan- lectin (MBL), H-ficolin and M-ficolin, three MBL-associated- isms or altered cells in the body. These molecules are found serine proteases (MASPs) and MBL-associated protein (MAp44), in complexes with MBL-associated serine proteases (MASP-1, and relation with functional genetic polymorphisms, in 130 MASP-2, or MASP-3) and with MBL-associated proteins (e.g., healthy children and upon intensive care unit (ICU) admission MAp-44). -
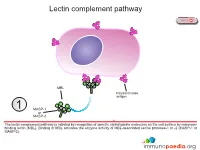
Lectin Complement Pathway
Lectin complement pathway NEXT MBL Polysaccharide antigen 1 MASP-1 or MASP-2 The lectin complement pathway is initiated by recognition of specific carbohydrate molecules on the cell surface by mannose- binding lectin (MBL). Binding of MBL activates the enzyme activity of MBL-associated serine protease-1 or -2 (MASP-1 or MASP-2). PREVIOUS NEXT C4b C4 C4a MASP-1 or 2 MASP-2 Activated MASP-1 or MASP-2 cleaves C4 complement protein into C4a that is released and C4b that binds to the cell surface. C4a functions as an anaphylatoxin that stimulates inflammation. PREVIOUS NEXT C3 C4b convertase {C2b C2b C2 MASP-1 or 3 C2a MASP-2 Activated MASP-1 or MASP-2 also cleaves C2 complement protein into C2a that is released and C2b that associates with C4b to constitute the C3 convertase enzyme system. PREVIOUS NEXT C3 C4b convertase {C2b C3 4 C3b C3a The C3 convertase cleaves C3 complement protein into C3a that is released and C3b that is required to constitute the C5 convertase enzyme system and also functions as an opsonin. PREVIOUS NEXT Opsonisation Click here for more detail C3b C4b C5 convertase { C2b C3b Opsonisation 5 C3b C3b associates with the C3 convertase to constitute the C5 convertase enzyme system. C3b also functions as an opsonin and binds to the surface of the target cell for enhanced detection by phagocytes expressing complement receptors. Follicular dendritic cells in germinal centres also express complement receptors which enhances capture of immune complexes for presentation to B lymphocytes. PREVIOUS NEXT C4b C5 convertase { C2b C3b C3b C5 6 C5a C5b The C5 convertase cleaves C5 complement protein into C5a that is released and C5b that recruits additional complement proteins invloved in the formation of the membrane attack complex (MAC). -

And Systems-Based Re-Engineering of Dendritic Cells with Non-Coding Rnas for Cancer Immunotherapy
Supplementary Materials Network- and systems-based re-engineering of dendritic cells with non-coding RNAs for cancer immunotherapy Xin Lai1,5,6,*, Florian S. Dreyer1,5,6, Martina Cantone1,5,6, Martin Eberhardt1,5,6, Kerstin F. Gerer2,5,6, Tanushree Jaitly1,5,6, Steffen Uebe3, Christopher Lischer1,5,6, Arif Ekici3, Jürgen Wittmann4, Hans-Martin Jäck4, Niels Schaft2,5,6, Jan Dörrie2,5,6, Julio Vera1,5,6,* 1. Laboratory of Systems Tumor Immunology, Department of Dermatology, Friedrich-Alexander- Universität Erlangen-Nürnberg (FAU) and Universitätsklinikum Erlangen, Erlangen, Germany 2. RNA Group, Department of Dermatology, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) and Universitätsklinikum Erlangen, Erlangen, Germany 3. Department of Human Genetics, Universitätsklinikum Erlangen, Erlangen, Germany 4. Division of Molecular Immunology, Department of Medicine 3, Universitätsklinikum Erlangen, Erlangen, Germany 5. Deutsches Zentrum Immuntherapie (DZI), Erlangen, Germany 6. Comprehensive Cancer Center (CCC) Erlangen, Erlangen, Germany * To whom correspondence should be addressed. Xin Lai, Tel: +49(0)91318545888, E-mail: xin.lai@uk- erlangen.de; Julio Vera, Tel: +49(0)91318545876, E-mail: [email protected]. Contents SUPPLEMENTARY TEXT ............................................................................................................................ 2 MicroRNA quantification ............................................................................................................................ 2 Reactome