Vacancies in Graphene: an Application of Adiabatic Quantum Optimization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anion Vacancies As a Source of Persistent Photoconductivity in II-VI and Chalcopyrite Semiconductors
PHYSICAL REVIEW B 72, 035215 ͑2005͒ Anion vacancies as a source of persistent photoconductivity in II-VI and chalcopyrite semiconductors Stephan Lany and Alex Zunger National Renewable Energy Laboratory, Golden, Colorado 80401, USA ͑Received 13 January 2005; revised manuscript received 12 April 2005; published 18 July 2005͒ Using first-principles electronic structure calculations we identify the anion vacancies in II-VI and chalcopy- rite Cu-III-VI2 semiconductors as a class of intrinsic defects that can exhibit metastable behavior. Specifically, ͑ ͒ we predict persistent electron photoconductivity n-type PPC caused by the oxygen vacancy VO in n-ZnO, originating from a metastable shallow donor state of VO. In contrast, we predict persistent hole photoconduc- ͑ ͒ tivity p-type PPC caused by the Se vacancy VSe in p-CuInSe2 and p-CuGaSe2. We find that VSe in the chalcopyrite materials is amphoteric having two “negative-U”-like transitions, i.e., a double-donor transition ͑2+/0͒ close to the valence band and a double-acceptor transition ͑0/2−͒ closer to the conduction band. We introduce a classification scheme that distinguishes two types of defects: type ␣, which have a defect-localized- state ͑DLS͒ in the band gap, and type , which have a resonant DLS within the host bands ͑e.g., the conduction band for donors͒. In the latter case, the introduced carriers ͑e.g., electrons͒ relax to the band edge where they can occupy a perturbed-host state. Type ␣ is nonconducting, whereas type  is conducting. We identify the neutral anion vacancy as type ␣ and the doubly positively charged vacancy as type . -

Study on Vacancy Defect Concentration and Hybrid Potential of Metal-Based Epitaxial Graphene with Temperature
Study on Vacancy Defect Concentration and Hybrid Potential of Metal-based Epitaxial Graphene with Temperature J H Gao, W Liu*, X X Ren and R L Zheng Engineering Research Center of New Energy Storage Devices and Applications, Chongqing Chongqing University of Arts and Sciences, Chongqing 402160, P. R.China Corresponding author and e-mail: W Liu, [email protected] Abstract. This paper investigates the variation of the vacancy defect concentration and hybrid potential of copper and nickel - based epitaxial graphene with temperature by considering the existence of vacancy defects. Moreover, we discuss the impact of atomic non - harmonic vibration on the metal - based epitaxial graphene. First, the concentration of vacancy defects increases nonlinearly with increasing temperature, and the changing rate of the vacancy defect concentration of the metal-based epitaxial graphene with temperature is smaller than that of graphene; Second, the hybrid potential increases with increasing temperature, but not much; Third, the hybrid potential is independent of temperature without considering the atomic non-harmonic vibration which has an important influence on the vacancy defect concentration and the hybrid potential. The higher the temperature is, the more significant the non-harmonic effects are. 1. Introduction Epitaxial graphene has attracted the attention of researchers worldwide for its application prospect [1-4]. Conductivity is one of the most widely used and most important properties. Beside the experimental research, many literatures have studied the conductivity of epitaxial graphene. In 2013, Z Z Alisultanov studied the electrical conductivity of electron gas on metal-based and semiconductor-based epitaxial graphene in Ref. [5]. In 2015, the variation of electrical conductivity with temperature and thickness of little layer graphene and graphene nanosheets were studied, indicating that the electrical conductivity gradually decreased with increasing temperature in Ref. -
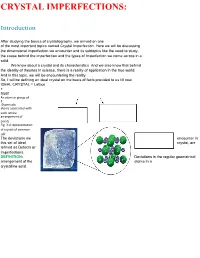
Crystal Imperfections-- Point, Line and Planar Defect
CRYSTAL IMPERFECTIONS: Introduction After studying the basics of crystallography, we arrived on one of the most important topics named Crystal Imperfection. Here we will be discussing the dimensional imperfection we encounter and its subtopics like the need to study, the cause behind the imperfection and the types of Imperfection we come across in a solid. We know about a crystal and its characteristics. And we also know that behind the ideality of theories in science, there is a reality of application in the true world. And in this topic, we will be encountering the reality. So, I will be defining an ideal crystal on the basis of facts provided to us till now. IDEAL CRYSTAL = Lattice + Motif An atom or group of 3 -D periodic atoms associated with each lattice arrangement of points Fig. 3-d representation of crystal of common salt The deviations we encounter in this set of ideal crystal, are termed as Defects or Imperfections. DEFINITION- Deviations in the regular geometrical arrangement of the atoms in a crystalline solid. Causes There can be a number of wanted and unwanted causes behind the defects that appear in a solid. Some are mentioned below: 1 .Deformation of solids (Alteration in size or shape of a body under the influence of mechanical forces) 2 3 4 .Rapid cooling from high temperature. .Crystallisation occurring at a differed rate. .High-energy radiation striking the solid. These reasons influence the mechanical, electrical, and optical behaviour of a crystal or a solid. Healthy Imperfections When we come across the word “Imperfection”, we assume that the thing for which this adjective is used is not perfect or worse than the perfect one. -

Vacancy Defect Positron Lifetimes in Strontium Titanate
1 Vacancy defect positron lifetimes in strontium titanate R.A. Mackie,1 S. Singh,1 J. Laverock, 2 S.B. Dugdale,2 and D.J. Keeble1,* 1Carnegie Laboratory of Physics, School of Engineering, Physics, and Mathematics, University of Dundee, Dundee DD1 4HN, UK 2 H.H. Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol BS8 1TL, UK *Corresponding author. Electronic address: [email protected] The results of positron annihilation lifetime spectroscopy measurements on undoped, electron irradiated, and Nb doped SrTiO3 single crystals are reported. Perfect lattice and vacancy defect positron lifetimes were calculated using two different first-principles schemes. The Sr vacancy defect related positron lifetime was obtained from measurements on Nb doped, electron irradiated, and vacuum annealed samples. Undoped crystals showed a defect lifetime component dominated by trapping to Ti vacancy related defects. 2 I. INTRODUCTION Vacancies are normally assumed to be the dominant point defects in perovskite oxide (ABO3) titanates, and often strongly influence the material properties such as electrical conductivity and domain stability and dynamics.1, 2 Strontium titanate is one of the most widely studied oxide materials, having a model cubic perovskite oxide structure at room temperature and exhibits an antiferrodistortive phase transition at 105 K. The material is a 0 3 wide band gap semiconductor (with Eg = 3.3 eV, ) but can be made a good conductor by 4 doping, for example, with Nb. Single crystal SrTiO3 substrates are readily available, and high quality epitaxial layers can be grown.5-7 The recent observation of two-dimensional 8 electron gas formation at the interface between SrTiO3 and LaAlO3, residing in the SrTiO3, has further demonstrated the importance of the material for future 9 multifunctional oxide electronic device structures. -

Defect and Metal Oxide Control of Schottky Barriers And
DEFECT AND METAL OXIDE CONTROL OF SCHOTTKY BARRIERS AND CHARGE TRANSPORT AT ZINC OXIDE INTERFACES DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Geoffrey Michael Reimbold Foster Graduate Program in Physics The Ohio State University 2018 Dissertation Committee: Professor Leonard J. Brillson Adviser Professor Thomas Lemberger Professor Ilya Gruzberg Professor Robert Perry Copyrighted by Geoffrey Michael Reimbold Foster 2018 ABSTRACT In recent years ZnO has received renewed interest due to its exciting semiconductor properties and remarkable ability to grow nanostructures. ZnO is a wide band gap semiconductor, allowing many potential future applications including electronic nanoscale devices, biosensors, blue/UV light emitters, and transparent conductors. There are many potential challenges that keep ZnO from reaching a full device potential. The biggest challenge is what the role native point defects play in the fabrication of high quality Ohmic and Schottky contacts. The following work examines the impact of these native point defects in the formation of Schottky barriers and charge transport at ZnO interfaces. We have used depth-resolved cathodoluminescence spectroscopy and nanoscale surface photovoltage spectroscopy to measure the dependence of native point defect energies and densities on Mg content, band gap, and lattice structure in non-polar, single- phase MgxZn1-xO (0<x<0.56) alloys grown by molecular beam epitaxy (MBE) on r-plane sapphire substrates. Based on this wide range of alloy compositions, we identified multiple deep level emissions due to zinc and oxygen vacancies whose densities exhibit a pronounced minimum at ~45% Mg corresponding to similar a and c parameter minima at ~52%. -

Understanding Defects in Germanium and Silicon for Optoelectronic Energy Conversion by Neil Sunil Patel B.S., Georgia Institute of Technology (2009)
Understanding Defects in Germanium and Silicon for Optoelectronic Energy Conversion by Neil Sunil Patel B.S., Georgia Institute of Technology (2009) Submitted to the Department of Materials Science and Engineering in partial fulfillment of the requirements for the degree of Doctor of Philosophy at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY June 2016 © Massachusetts Institute of Technology 2016. All rights reserved. Author.......................................................................... Department of Materials Science and Engineering May 17, 2016 Certified by . Lionel C. Kimerling Thomas Lord Professor of Materials Science and Engineering Thesis Supervisor Certified by . Anuradha M. Agarwal Principal Research Scientist, Materials Processing Center Thesis Supervisor Acceptedby..................................................................... Donald Sadoway Chairman, Department Committee on Graduate Theses Understanding Defects in Germanium and Silicon for Optoelectronic Energy Conversion by Neil Sunil Patel Submitted to the Department of Materials Science and Engineering on May 17, 2016, in partial fulfillment of the requirements for the degree of Doctor of Philosophy Abstract This thesis explores bulk and interface defects in germanium (Ge) and silicon (Si) with a focus on understanding the impact defect related bandgap states will have on optoelectronic applications. Optoelectronic devices are minority carrier devices and are particularly sensi- tive to defect states which can drastically reduce carrier lifetimes in small concentrations. We performed a study of defect states in Sb-doped germanium by generation of defects via irradiation followed by subsequent characterization of electronic properties via deep- level transient spectroscopy (DLTS). Cobalt-60 gamma rays were used to generate isolated vacancies and interstitials which diffuse and react with impurities in the material toform four defect states (E37,E30,E22, and E21) in the upper half of the bandgap. -

Defects and Disorders in Semiconductors
1 Defects and Disorders in Semiconductors Jermell Powell, Electrical and Computer Engineering position, and interstitials, in which an atom is located in Abstract— Various defects and disorders in between lattice sites. Also, when dealing with doping or semiconductors will be described, along with their causes fabrication, one can encounter impurities. These atoms which and consequences. Determination and classification of are different than the original material may be substitutional defects will follow. Many defects are general; however, defects, when they replace an intended atom at a lattice specific materials will be examined. position, or interstitial impurities [4]. Figure 1 provides examples for four of the previously stated defects. Index Terms — Defect Density, Point defects, Vacancy, Silicon I. INTRODUCTION HERE is an abundance of previous research that has T determined and categorized semiconductor defects. The simplified case is to assume that defects occur randomly over a silicon slice. Further in-depth analysis has shown that this simplification is not realistic, however [1]. Most faults have been shown to lie at the material’s edge, due to thermal and mechanical stress. And while classically, fault densities were considered with respect to radial variation, further attention has been given to angular deviation. Both variations have been studied for analysis of defect phenomena, including defect clusters and slip lines. Fig 1. Common point defects in semiconductors. Substitutional and interstitial defects involve a separate II. DEFECT TYPES ion, whereas self interstitials are due to an original atom. The categories of defective circuits are as follows: A) Local faults initially in the material; Area defects are thought of as extended point defects, B) Local faults created during fabrication; particularly epitaxial mounds. -

Ab Initio Electron-Defect Interactions Using Wannier Functions ✉ I-Te Lu1, Jinsoo Park1, Jin-Jian Zhou1 and Marco Bernardi1
www.nature.com/npjcompumats ARTICLE OPEN Ab initio electron-defect interactions using Wannier functions ✉ I-Te Lu1, Jinsoo Park1, Jin-Jian Zhou1 and Marco Bernardi1 Computing electron–defect (e–d) interactions from first principles has remained impractical due to computational cost. Here we develop an interpolation scheme based on maximally localized Wannier functions (WFs) to efficiently compute e–d interaction matrix elements. The interpolated matrix elements can accurately reproduce those computed directly without interpolation and the approach can significantly speed up calculations of e–d relaxation times and defect-limited charge transport. We show example calculations of neutral vacancy defects in silicon and copper, for which we compute the e–d relaxation times on fine uniform and random Brillouin zone grids (and for copper, directly on the Fermi surface), as well as the defect-limited resistivity at low temperature. Our interpolation approach opens doors for atomistic calculations of charge carrier dynamics in the presence of defects. npj Computational Materials (2020) 6:17 ; https://doi.org/10.1038/s41524-020-0284-y INTRODUCTION an open problem. Interpolating the interaction matrix elements to The interactions between electrons and defects control charge uniform, random, or importance-sampling fine BZ grids is key to 25,28 and spin transport at low temperature, and give rise to a range of systematically converging the RTs and transport properties , 1234567890():,; quantum transport phenomena1–5. Understanding such and it has been an important development in first-principles – electron–defect (e–d) interactions from first principles can provide calculations of e ph interactions and phonon-limited charge microscopic insight into carrier dynamics in materials in the transport. -

Effects of Monovacancy and Divacancies on Hydrogen Solubility
materials Article Effects of Monovacancy and Divacancies on Hydrogen Solubility, Trapping and Diffusion Behaviors in fcc-Pd by First Principles 1, 2, 3 3 4 4 Bao-Long Ma y , Yi-Yuan Wu y, Yan-Hui Guo , Wen Yin , Qin Zhan , Hong-Guang Yang , Sheng Wang 1,* and Bao-Tian Wang 3,5,* 1 Department of Nuclear Science and Technology, School of Energy and Power Engineering, Xi’an Jiaotong University, Xi’an 710049, China; [email protected] 2 Engineering Research Center of Nuclear Technology Application, Ministry of Education, East China University of Technology, Nanchang 330013, China; [email protected] 3 Spallation Neutron Source Science Center, Institute of High Energy Physics, Chinese Academy of Sciences, Dongguan 523803, China; [email protected] (Y.-H.G.); [email protected] (W.Y.) 4 Department of Reactor Engineering Research & Design, China Institute of Atomic Energy, Beijing 102413, China; [email protected] (Q.Z.); [email protected] (H.-G.Y.) 5 Innovation Center of Extreme Optics, Shanxi University, Taiyuan 030006, China * Correspondence: [email protected] (S.W.); [email protected] (B.-T.W.) These authors contributed equally to this work. y Received: 11 October 2020; Accepted: 28 October 2020; Published: 30 October 2020 Abstract: The hydrogen blistering phenomenon is one of the key issues for the target station of the accelerator-based neutron source. In the present study, the effect of monovacancies and divacancies defects on the solution, clustering and diffusion behaviors of H impurity in fcc-Pd were studied through first principles calculations. Our calculations prove that vacancies behave as an effective sink for H impurities. -

Defect Studies in Metals, Alloys, and Oxides by Positron Annihilation Spectroscopy and Related Techniques
DEFECT STUDIES IN METALS, ALLOYS, AND OXIDES BY POSITRON ANNIHILATION SPECTROSCOPY AND RELATED TECHNIQUES Sahil Agarwal A Dissertation Submitted to the Graduate College of Bowling Green State University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY August 2021 Committee: Farida Selim, Advisor Abby Braden Graduate Faculty Representative Alexander Tarnovsky Marco Nardone © 2021 Sahil Agarwal All Rights Reserved iii ABSTRACT Farida Selim Advisor Positrons administer a unique non-destructive approach to probe materials with atomic- scale sensitivity and provide reliable information about the nature and size of defects. This study reflects on the powerful capabilities of positron annihilation spectroscopy (PAS) to characterize point defects at atomic-scale, which can be crucial in the development of relevant material for a wide range of applications like nuclear reactors, medical sciences, optoelectronic devices, nanotechnology, polymers etc. The work presented in this dissertation aims to gain a fundamental understanding of the defect structures in three unique material systems: Fe metal, Fe-Cr alloy, and Ce:YAG oxide by applying the methodology and concepts of PAS. A wide range of other complementary characterization techniques has also been employed to enhance the understanding. The depth-resolved PAS was used to identify vacancy clusters in ion irradiated Fe and measure their density as a function of depth. PAS measurements uncovered the structure of vacancy clusters and the change in their size and density with irradiation dose. Combining with TEM measurements led to discovering a novel mechanism for the interaction of cascade damage with voids in ion-irradiated materials. The effect of Cr alloying on the formation and evolution of atomic size clusters induced by ion irradiation in Fe-Cr alloys was also investigated using depth-resolved PAS measurements. -

Atomic Vacancy Defect, Frenkel Defect and Transition Metals (Sc, V, Zr) Doping in Ti4n3 Mxene Nanosheet: a First-Principles Investigation
applied sciences Article Atomic Vacancy Defect, Frenkel Defect and Transition Metals (Sc, V, Zr) Doping in Ti4N3 MXene Nanosheet: A First-Principles Investigation Tingyan Zhou 1,2, Wan Zhao 3, Kun Yang 2, Qian Yao 1, Yangjun Li 2, Bo Wu 2,4 and Jun Liu 1,* 1 School of Science, Chongqing University of Posts and Telecommunications, Chongqing 400065, China; [email protected] (T.Z.); [email protected] (Q.Y.) 2 School of Physics and Electronic Science, Zunyi Normal University, Zunyi 563006, China; [email protected] (K.Y.); [email protected] (Y.L.); [email protected] (B.W.) 3 School of Physics, Beijing Institute of Technology, Beijing 100081, China; [email protected] 4 School of Marine Science and Technology, Northwestern Polytechnical University, Xi’an 710072, China * Correspondence: [email protected] Received: 8 March 2020; Accepted: 31 March 2020; Published: 3 April 2020 Abstract: Using first-principles calculations based on the density functional theory, the effects of atomic vacancy defect, Frenkel-type defect and transition metal Z (Z = Sc, V and Zr) doping on magnetic and electric properties of the Ti4N3 MXene nanosheet were investigated comprehensively. The surface Ti and subsurface N atomic vacancies are both energetically stable based on the calculated binding energy and formation energy. In addition, the former appears easier than the latter. They can both enhance the magnetism of the Ti4N3 nanosheet. For atom-swapped disordering, the surface Ti-N swapped disordering is unstable, and then the Frenkel-type defect will happen. In the Frenkel-type defect system, the total magnetic moment decreases due to the enhancement of indirect magnetic exchange between surface Ti atoms bridged by the N atom. -

Calculation of the Properties of Vacancies and Interstitials, Skyland
UNITED STATES DEPARTMENT OF COMMERCE Alexander B. Trowbridge, Secretary • NATIONAL BUREAU OF STANDARDS • A. V. Astin, Director Calculation of the Properties of Vacancies and Interstitials Proceedings of a Conference Shenandoah National Park, Va. May 1-5, 1966 Supported in part by the Advanced Research Projects Agency National Bureau of Standards Miscellaneous Publication 287 Issued November 17, 1966 For sale by the Superintendent of Documents, U.S. Government Printing Office. Washington, D.C 20402 Price $2.50 Abstract This is the Proceedings of a Conference on the Calculation of the Properties of Vacancies and Interstitials, Skyland. Virginia, USA, held on May 1-4, 1966. The Conference dealt with the theory and techniques of calculation of the properties of point defects in metallic and nonmetallic crystals. The contributed and invited papers divided about evenly among three major topics: (1) static-lattice calculations of the energies and configurations of simple vacancies and interstitials in, mainly, metals and ionic crystals; (2) electronic states at and near point defects in metals, rare gas solids, and insula- tors (/-centers, electron traps); and (3) vibrational states at point defects. The report of a panel dis- cussion on each topic is also included. The emphasis is on the theory of the properties of isolated, simple defects rather than on the statistical properties of defect assemblies. The Conference attempted to examine the point defect theory and calculations critically, from the standpoint of general theory, rather than simply compare results with experiment. Key Words: Calculations, electronic states, energies of formation, energies of motion, interstitials, point defects, theory, vacancies, vibrational states.