The University of Chicago Genetic Services Laboratories Labolaboratories 5841 S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Ion Channels
UC Davis UC Davis Previously Published Works Title THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Permalink https://escholarship.org/uc/item/1442g5hg Journal British journal of pharmacology, 176 Suppl 1(S1) ISSN 0007-1188 Authors Alexander, Stephen PH Mathie, Alistair Peters, John A et al. Publication Date 2019-12-01 DOI 10.1111/bph.14749 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology (2019) 176, S142–S228 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels Stephen PH Alexander1 , Alistair Mathie2 ,JohnAPeters3 , Emma L Veale2 , Jörg Striessnig4 , Eamonn Kelly5, Jane F Armstrong6 , Elena Faccenda6 ,SimonDHarding6 ,AdamJPawson6 , Joanna L Sharman6 , Christopher Southan6 , Jamie A Davies6 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 3Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 4Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, A-6020 Innsbruck, Austria 5School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 6Centre for Discovery Brain Science, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. -

Modulation of Voltage-Gated Potassium Channels by Phosphatidylinositol-4,5-Bisphosphate Marina Kasimova
Modulation of voltage-gated potassium channels by phosphatidylinositol-4,5-bisphosphate Marina Kasimova To cite this version: Marina Kasimova. Modulation of voltage-gated potassium channels by phosphatidylinositol-4,5- bisphosphate. Other. Université de Lorraine, 2014. English. NNT : 2014LORR0204. tel-01751176 HAL Id: tel-01751176 https://hal.univ-lorraine.fr/tel-01751176 Submitted on 29 Mar 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. AVERTISSEMENT Ce document est le fruit d'un long travail approuvé par le jury de soutenance et mis à disposition de l'ensemble de la communauté universitaire élargie. Il est soumis à la propriété intellectuelle de l'auteur. Ceci implique une obligation de citation et de référencement lors de l’utilisation de ce document. D'autre part, toute contrefaçon, plagiat, reproduction illicite encourt une poursuite pénale. Contact : [email protected] LIENS Code de la Propriété Intellectuelle. articles L 122. 4 Code de la Propriété Intellectuelle. articles L 335.2- L 335.10 http://www.cfcopies.com/V2/leg/leg_droi.php -

Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery
fphar-07-00121 May 7, 2016 Time: 11:45 # 1 REVIEW published: 10 May 2016 doi: 10.3389/fphar.2016.00121 Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery Paola Imbrici1*, Antonella Liantonio1, Giulia M. Camerino1, Michela De Bellis1, Claudia Camerino2, Antonietta Mele1, Arcangela Giustino3, Sabata Pierno1, Annamaria De Luca1, Domenico Tricarico1, Jean-Francois Desaphy3 and Diana Conte1 1 Department of Pharmacy – Drug Sciences, University of Bari “Aldo Moro”, Bari, Italy, 2 Department of Basic Medical Sciences, Neurosciences and Sense Organs, University of Bari “Aldo Moro”, Bari, Italy, 3 Department of Biomedical Sciences and Human Oncology, University of Bari “Aldo Moro”, Bari, Italy In the human genome more than 400 genes encode ion channels, which are transmembrane proteins mediating ion fluxes across membranes. Being expressed in all cell types, they are involved in almost all physiological processes, including sense perception, neurotransmission, muscle contraction, secretion, immune response, cell proliferation, and differentiation. Due to the widespread tissue distribution of ion channels and their physiological functions, mutations in genes encoding ion channel subunits, or their interacting proteins, are responsible for inherited ion channelopathies. These diseases can range from common to very rare disorders and their severity can be mild, Edited by: disabling, or life-threatening. In spite of this, ion channels are the primary target of only Maria Cristina D’Adamo, University of Perugia, Italy about 5% of the marketed drugs suggesting their potential in drug discovery. The current Reviewed by: review summarizes the therapeutic management of the principal ion channelopathies Mirko Baruscotti, of central and peripheral nervous system, heart, kidney, bone, skeletal muscle and University of Milano, Italy Adrien Moreau, pancreas, resulting from mutations in calcium, sodium, potassium, and chloride ion Institut Neuromyogene – École channels. -

Molecular Identi¢Cation and Characterisation of the Human Eag2
FEBS Letters 524 (2002) 204^210 FEBS 26330 View metadata, citation and similar papers at core.ac.uk brought to you by CORE Molecular identi¢cation and characterisation of the humanprovided eag2 by Elsevier - Publisher Connector potassium channel M. Ju, D. Wrayà School of Biomedical Sciences, University of Leeds, Leeds LS2 9JT, UK Received 27 May 2002; revised 25June 2002; accepted 28 June 2002 First published online 9 July 2002 Edited by Maurice Montal without inactivation; the (slow) activation kinetics depend on Abstract We report the molecular cloning from foetal brain of the human potassium channel heag2. The cDNA encodes a pro- holding potential (‘Cole^Moore shift’) and extracellular mag- tein of 988 amino acids, 73%identical to heag1. Heag2 is ex- nesium, while the channel can be blocked by intracellular pressed in the brain, but is also found in a range of tissues calcium. The rat eag channels are expressed predominantly including skeletal muscle. In oocytes, the channel is a non-in- in neural tissue; cell cycle-speci¢c expression of eag1 has activating outward recti¢er, with dependence of activation rate been demonstrated that may be of great importance in cellular on holding potential. Compared with heag1, the conductance^ development and tumour growth. Finally, members of the elk voltage curve for heag2 was shifted to the left, the voltage subfamily [3] show electrophysiological features like eag (e.g. sensitivity was less, activation kinetics were di¡erent, and the elk1), or features intermediate between eag and erg (e.g. elk2), sensitivity to terfenadine was lower. The heag2 channel may though they do not show the Cole^Moore shift [3]. -
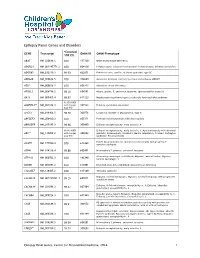
Epilepsy Panel Genes and Disorders
Epilepsy Panel Genes and Disorders *Covered GENE Transcript OMIM ID OMIM Phenotype 10X (%) ABAT NM_020686.5 100 137150 GABA-transaminase deficiency ADGRG1 NM_001145771.2 100 604110 Polymicrogyria, bilateral frontoparietal; Polymicrogyria, bilateral perisylvian ADGRV1 NM_032119.3 99.93 602851 Febrile seizures, familial, 4; Usher syndrome, type 2C ADRA2B NM_000682.5 100 104260 Autosomal dominant cortical myoclonus and epilepsy (ADCME) ADSL NM_000026.2 100 608222 Adenylosuccinase deficiency AFG3L2 NM_006796.2 98.16 604581 Ataxia, spastic, 5, autosomal recessive; Spinocerebellar ataxia 28 AKT3 NM_005465.4 99.97 611223 Megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome 93.05 (100% ALDH7A1** NM_001182.3 with Sanger 107323 Epilepsy, pyridoxine-dependent Gap fill) ALG13 NM_018466.5 98.76 300776 Congenital disorder of glycosylation, type Is ARFGEF2 NM_006420.2 100 605371 Periventricular heterotopia with microcephaly ARHGEF9 NM_015185.3 99.96 300430 Epileptic encephalopathy, early infantile, 8 98.96 (100% Epileptic encephalopathy, early infantile, 1; Hydranencephaly with abnormal ARX** NM_139058.2 with Sanger 300382 genitalia; Lissencephaly, X-linked 2; Mental retardation, X-linked; Partington Gap fill) syndrome; Proud syndrome ASAH1 NM_177924.3 613468 Farber lipogranulomatosis; Spinal muscular atrophy with progressive 100 myoclonic epilepsy ASPM NM_018136.4 99.88 605481 Microcephaly 5, primary, autosomal recessive ATP1A2 NM_000702.3 182340 Alternating hemiplegia of childhood; Migraine, familial basilar; Migraine, 100 familial hemiplegic, -

Gene List of the Targeted NGS MCD and CCA Gene Panel AKT3,ALX1
Gene List of the targeted NGS MCD and CCA gene panel AKT3,ALX1,ALX3,ALX4,AMPD2,ARFGEF2,ARID1B,ARX,ASPM,ATR,ATRX,B3GALTL,BRPF1,c12orf57,C6orf70,CASK,CCND2,CDK5RAP2,CDON,C ENPJ,CEP170,CHMP1A,COL4A1,CREBBP,CYP11A1,DCHS1,DCLK1,DCX,DHCR24,DHCR7,DIS3L2,DISC1,DISP1,DLL1,DMRTA2,DYNC1H1,DYRK1 A,EARS2,EFNB1,EMX1,EOMES,EP300,ERBB4,ERMARD,EXOSC3,FAM36A,FGF8,FGFR1,FGFR2,FLNA,FOXC1,FOXG1,FOXH1,FZD10,GLI2,GLI3,GP R56,GPSM2,HCCS,HESX1,HNRNPU,IGBP1,IGFBP1,ISPD,ITPA,KAL1,KAT6B,KATNB1,KIAA1279,KIF14,KIF1A,KIF1B,KIF21A,KIF2A,KIF5C,KIF7,L1 CAM,LAMB1,LAMC3,LRP2,MCPH1,MED12,MID1,NDE1,NFIB,NPC1,NR2F1,NSD1,NTRK1,NTRK3,OCEL1,OPA1,OTX2,PAFAH1B1,PAX6,PEX1,PHF1 0,PIK3R2,POLR3A,POLR3B,POMT1,POMT2,PTCH1,PTPRS,PYCR1,RAB3GAP1,RARS2,RELN,RFX3,ROBO1,ROBO3,RPS6KA3,RTTN,SATB2,SEPSEC S,SHH,SIX3,SLC12A6,SOX2,SPOCK1,SRPX2,TBCD,TBCE,TCF4,TDGF1,TEAD1,THBS2,TMEM5,TSC1,TSC2,TSEN15,TSEN2,TSEN34,TSEN54,TUBA1 A,TUBA8,TUBB,TUBB2A,TUBB2B,TUBB3,TUBB4A,TUBG1,VAX1,VRK1,WDR47,WDR62,ZBTB18,ZEB2,ZIC2. Gene List of the targeted NGS epilepsy gene panel AARS, ADGRV1, ADRA2B, ADSL, ALDH4A1, ALDH7A1, ALG13, ALPL, ARHGEF15, ARHGEF9, ARX, ASAH1, ATP1A2, ATP1A3, BRD2, CACNA1A, CACNA1H, CACNA2D2, CACNB4, CBL, CDKL5, CERS1, CHD2, CHRNA2, CHRNA4, CHRNB2, CLCN2, CLCN4, CLN8, CLTC, CNKSR2, CNTNAP2, CPA6, CPLX1, CSNK1G1, CSNK2B, CTNND2, DEPDC5, DHDDS, DNM1, DOCK7, DYNC1H1, EEF1A2, EFHC1, EIF2S3, EMC1, EPM2A, FASN, FLNA, FOXG1, GABBR2, GABRA1, GABRA2, GABRA3, GABRB2, GABRB3, GABRD, GABRG2, GAL, GNAO1, GOSR2, GRIA1, GRIN1, GRIN2A, GRIN2B, HCN1, HCN4, HDAC4, HNRNPU, IDH3A, IQSEC2, JRK, KCNA1, KCNA2, KCNB1, -

Pharmacological Target-Focused Transcriptomic Analysis of Native Vs Cultured Human and Mouse Dorsal Root Ganglia Andi Wangzhoua, Lisa A
Research Paper Pharmacological target-focused transcriptomic analysis of native vs cultured human and mouse dorsal root ganglia Andi Wangzhoua, Lisa A. McIlvriedb, Candler Paigea, Paulino Barragan-Iglesiasa, Stephanie Shiersa, Ayesha Ahmada, Carolyn A. Guzmana, Gregory Dussora, Pradipta R. Raya,*, Robert W. Gereau IVb, Theodore J. Pricea Abstract Dorsal root ganglion (DRG) neurons detect sensory inputs and are crucial for pain processing. They are often studied in vitro as dissociated cell cultures with the assumption that this reasonably represents in vivo conditions. However, to the best of our knowledge, no study has directly compared genome-wide transcriptomes of DRG tissue in vivo versus in vitro or between laboratories and culturing protocols. Comparing RNA sequencing-based transcriptomes of native to cultured (4 days in vitro) human or mouse DRG, we found that the overall expression levels of many ion channels and G-protein–coupled receptors specifically expressed in neurons are markedly lower although still expressed in culture. This suggests that most pharmacological targets expressed in vivo are present under the condition of dissociated cell culture, but with changes in expression levels. The reduced relative expression for neuronal genes in human DRG cultures is likely accounted for by increased expression of genes in fibroblast-like and other proliferating cells, consistent with their mitotic status in these cultures. We found that the expression of a subset of genes typically expressed in neurons increased in human and mouse DRG cultures relative to the intact ganglion, including genes associated with nerve injury or inflammation in preclinical models such as BDNF, MMP9, GAL,andATF3. We also found a striking upregulation of a number of inflammation- associated genes in DRG cultures, although many were different between mouse and human. -

Genetic Variations Associated with Pharmacoresistant Epilepsy (Review)
MOLECULAR MEDICINE REPORTS 21: 1685-1701, 2020 Genetic variations associated with pharmacoresistant epilepsy (Review) NOEMÍ CÁRDENAS-RODRÍGUEZ1*, LILIANA CARMONA-APARICIO1*, DIANA L. PÉREZ-LOZANO1,2, DANIEL ORTEGA-CUELLAR3, SAÚL GÓMEZ-MANZO4 and IVÁN IGNACIO-MEJÍA5,6 1Laboratory of Neuroscience, National Institute of Pediatrics, Ministry of Health, Coyoacán, Mexico City 04530; 2Department of Postgraduate of Medical, Dental and Health Sciences, Clinical Experimental Health Research, National Autonomous University of Mexico, Mexico City 04510; Laboratories of 3Experimental Nutrition and 4Genetic Biochemistry, National Institute of Pediatrics, Ministry of Health, Coyoacán, Mexico City 04530; 5Laboratory of Translational Medicine, Military School of Health Graduates, Lomas de Sotelo, Militar, Mexico City 11200; 6Section of Research and Graduate Studies, Superior School of Medicine, National Polytechnic Institute, Mexico City 11340, Mexico Received August 28, 2019; Accepted January 16, 2020 DOI: 10.3892/mmr.2020.10999 Abstract. Epilepsy is a common, serious neurological disorder the pathophysiological processes that underlie this common worldwide. Although this disease can be successfully treated human neurological disease. in most cases, not all patients respond favorably to medical treatments, which can lead to pharmacoresistant epilepsy. Drug-resistant epilepsy can be caused by a number of mecha- Contents nisms that may involve environmental and genetic factors, as well as disease- and drug-related factors. In recent years, 1. Introduction numerous studies have demonstrated that genetic variation is 2. Pharmacoresistant epilepsy involved in the drug resistance of epilepsy, especially genetic 3. Genetic variations associated with pharmacoresistant variations found in drug resistance-related genes, including epilepsy the voltage-dependent sodium and potassium channels genes, 4. The role of genetic variants in the diagnosis and treatment and the metabolizer of endogenous and xenobiotic substances of pharmacoresistant epilepsy genes. -

PDF, Also Known As Version of Record
Molecular basis of potassium channels in pancreatic duct epithelial cells Hayashi, M.; Novak, Ivana Published in: Channels (Austin) DOI: 10.4161/chan.26100 Publication date: 2013 Document version Publisher's PDF, also known as Version of record Citation for published version (APA): Hayashi, M., & Novak, I. (2013). Molecular basis of potassium channels in pancreatic duct epithelial cells. Channels (Austin), 7(6), 432-441. https://doi.org/10.4161/chan.26100 Download date: 23. sep.. 2021 PAPER TYPE Channels 7:6, 432–441; November/December 2013; © 2013 Landes Bioscience Molecular basis of potassium channels in pancreatic duct epithelial cells Mikio Hayashi†,* and Ivana Novak* Department of Biology; University of Copenhagen; Copenhagen, Denmark; †Current affiliation: Department of Physiology; Kansai Medical University; Hirakata, Japan Keywords: cancer, EAG2, epithelia, HERG, pancreas, SK4, Slack, Slick, Slo1, TASK-2 Abbreviations: AKAP, A-kinase anchoring protein; BK, voltage – and Ca2+-dependent maxi-K+; BxPC3, human pancreas adenocarcinoma cell line; Capan-1, human pancreas adenocarcinoma cell line; CFPAC-1, human cystic fibrosis pancreatic adenocarcinoma cell line; CFTR, cystic fibrosis transmembrane conductance regulator; DC-EBIO, 5,6-dichloro-1-ethyl-1,3- dihydro-2H-benzimidazole-2-one; DHS-I, dehydrosoyasaponin I; DIDS, 4,4’-diisothiocyanatostilbene-2,2’-disulfonic acid; E-4031, N-[4-[1-[2-(6-methylpyridin-2-yl)ethyl]piperidine-4-carbonyl]phenyl] methanesulfonamide; EAG, ether-à-go-go gene; 1-EBIO, 1-ethyl-2-benzimidazolinone; HERG, -

Multistate Structural Modeling and Voltage-Clamp Analysis of Epilepsy/Autism Mutation Kv10.2–R327H Demonstrate the Role Of
16586 • The Journal of Neuroscience, October 16, 2013 • 33(42):16586–16593 Neurobiology of Disease Multistate Structural Modeling and Voltage-Clamp Analysis of Epilepsy/Autism Mutation Kv10.2–R327H Demonstrate the Role of This Residue in Stabilizing the Channel Closed State Yang Yang,1,2,3* Dmytro V. Vasylyev,1,2,3* Fadia Dib-Hajj,1,2,3 Krishna R. Veeramah,4 Michael F. Hammer,4 Sulayman D. Dib-Hajj,1,2,3 and Stephen G. Waxman1,2,3 1Department of Neurology and 2Center for Neuroscience and Regeneration Research, Yale University School of Medicine, New Haven, Connecticut 06510, 3Rehabilitation Research Center, Veterans Affairs Connecticut Healthcare System, West Haven, Connecticut 06516, and 4Arizona Research Laboratories Division of Biotechnology, University of Arizona, Tucson, Arizona 85721 Voltage-gated potassium channel Kv10.2 (KCNH5) is expressed in the nervous system, but its functions and involvement in human disease are poorly understood. We studied a human Kv10.2 channel mutation (R327H) recently identified in a child with epileptic encephalopathy and autistic features. Using multistate structural modeling, we demonstrate that the Arg327 residue in the S4 helix of voltage-sensingdomainhasstrongionicinteractionswithnegativelychargedresidueswithintheS1–S3helicesintheresting(closed)and early-activation state but not in the late-activation and fully-activated (open) state. The R327H mutation weakens ionic interactions between residue 327 and these negatively charged residues, thus favoring channel opening. Voltage-clamp analysis showed a strong hyperpolarizing(ϳ70mV)shiftofvoltagedependenceofactivationandanaccelerationofactivation.Ourresultsdemonstratethecritical role of the Arg327 residue in stabilizing the channel closed state and explicate for the first time the structural and functional change of a Kv10.2 channel mutation associated with neurological disease.