Methanogen Oxygen Stress: an Investigation of the Superoxide Dismutase Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Bacteria and Archaea in the Deep-Sea Low-Temperature Hydrothermal Sulfide Chimney of the Northeastern Pacific Ocean
African Journal of Biotechnology Vol. 11(2), pp. 337-345, 5 January, 2012 Available online at http://www.academicjournals.org/AJB DOI: 10.5897/AJB11.2692 ISSN 1684–5315 © 2012 Academic Journals Full Length Research Paper Diversity of bacteria and archaea in the deep-sea low-temperature hydrothermal sulfide chimney of the Northeastern Pacific Ocean Xia Ding1*, Xiao-Jue Peng1#, Xiao-Tong Peng2 and Huai-Yang Zhou3 1College of Life Sciences and Key Laboratory of Poyang Lake Environment and Resource Utilization, Ministry of Education, Nanchang University, Nanchang 330031, China. 2Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou 510640, China. 3National Key Laboratory of Marine Geology, Tongji University, Shanghai 200092, China. Accepted 4 November, 2011 Our knowledge of the diversity and role of hydrothermal vents microorganisms has considerably expanded over the past decade, while little is known about the diversity of microorganisms in low-temperature hydrothermal sulfide chimney. In this study, denaturing gradient gel electrophoresis (DGGE) and 16S rDNA sequencing were used to examine the abundance and diversity of microorganisms from the exterior to the interior of the deep sea low-temperature hydrothermal sulfide chimney of the Northeastern Pacific Ocean. DGGE profiles revealed that both bacteria and archaea could be examined in all three zones of the chimney wall and the compositions of microbial communities within different zones were vastly different. Overall, for archaea, cell abundance was greatest in the outermost zone of the chimney wall. For bacteria, there was no significant difference in cell abundance among three zones. In addition, phylogenetic analysis revealed that Verrucomicrobia and Deltaproteobacteria were the predominant bacterial members in exterior zone, beta Proteobacteria were the dominant members in middle zone, and Bacillus were the abundant microorganisms in interior zone. -

FORMATION of HABITABLE WORLDS and FATE of HABITABLE ENVIRONMENTS 6:00 P.M
45th Lunar and Planetary Science Conference (2014) sess726.pdf Thursday, March 20, 2014 [R726] POSTER SESSION: FORMATION OF HABITABLE WORLDS AND FATE OF HABITABLE ENVIRONMENTS 6:00 p.m. Town Center Exhibit Area Johnson T. A. Park A. Hand K. P. POSTER LOCATION #473 The Workman-Reynolds Effect: An Investigation of the Ice-Water Interface of Dilute Salt Solutions [#1672] We report on experiments on the voltage potential that results from rapidly freezing dilute aqueous solutions. These results have application to icy satellites. Taylor A. R. Olsen A. A. Hausrath E. M. POSTER LOCATION #474 Serpentinite Dissolution: An Analog to Mantle-Ocean Interaction on Europa [#1903] Laboratory-based dissolution experiments between serpentinite rock and a variety of acids represent mantle-ocean interaction on the jovian moon, Europa. Hausrath E. M. Adcock C. T. Elwood Madden M. E. Gainey S. R. Olsen A. A. et al. POSTER LOCATION #475 Using Geochemical Kinetics to Interpret Potential Habitability [#2376] Geochemical kinetics can help shed light on factors affecting habitability, including water, release of nutrients, redox, pH, temperature, and ionic strength. Som S. M. Fristad K. E. Hoehler T. M. POSTER LOCATION #476 An Integrative Approach to Assessing Habitability of H2 Metabolisms in Hydrothermal Springs [#2828] We present an ongoing project that surveys H2 from springs sourced in rocks of varying silica content and in parallel investigate habitability numerically. Djordjevic S. Mickol R. L. Kral T. A. POSTER LOCATION #477 Simulating Martian Conditions: Methanogen Survivability During Freeze-Thaw Cycles [#2539] Methanogens are obligate anaerobes that tolerate a wide range of conditions. It is proposed that these Archaea are able to persist in a martian environment. -

Susceptibility of Archaea to Antimicrobial Agents: Applications to Clinical Microbiology
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REVIEW 10.1111/j.1469-0691.2012.03913.x Susceptibility of archaea to antimicrobial agents: applications to clinical microbiology S. Khelaifia and M. Drancourt Unite´ de Recherche sur les Maladies Infectieuses et Tropicales Emergentes, UMR CNRS 6236 IRD 3R198, Me´diterrane´e Infection, Faculte´ de Me´decine, Aix-marseille-Universite´, Marseille, France Abstract We herein review the state of knowledge regarding the in vitro and in vivo susceptibility of archaea to antimicrobial agents, including some new molecules. Indeed, some archaea colonizing the human microbiota have been implicated in diseases such as periodontopathy. Archaea are characterized by their broad-spectrum resistance to antimicrobial agents. In particular, their cell wall lacks peptidoglycan, making them resistant to antimicrobial agents interfering with peptidoglycan biosynthesis. Archaea are, however, susceptible to the pro- tein synthesis inhibitor fusidic acid and imidazole derivatives. Also, squalamine, an antimicrobial agent acting on the cell wall, proved effective against human methanogenic archaea. In vitro susceptibility data could be used to design protocols for the decontamination of complex microbiota and the selective isolation of archaea in anaerobic culture. Keywords: Antimicrobial agent, archaea, methanogenic archaea, microbiota, susceptibility testing Article published online: 23 May 2012 Clin Microbiol Infect 2012; 18: 841–848 Corresponding author: M. Drancourt, Unite´ des Rickettsies, Fa- culte´ de Me´decine, 27, Boulevard Jean Moulin-Cedex 5, France E-mail: [email protected] Methanogenic archaea (herein referred to as methano- Introduction gens) are the sole organisms producing methane from H2 +CO2 [6]. -

DOGAMI Open-File Report O-86-06, the State of Scientific
"ABLE OF CONTENTS Page INTRODUCTION ..~**********..~...~*~~.~...~~~~1 GORDA RIDGE LEASE AREA ....................... 2 RELATED STUDIES IN THE NORTH PACIFIC .+,...,. 5 BYDROTHERMAL TEXTS ........................... 9 34T.4 GAPS ................................... r6 ACKNOWLEDGEMENT ............................. I8 APPENDIX 1. Species found on the Gorda Ridge or within the lease area . .. .. .. .. .. 36 RPPENDiX 2. Species found outside the lease area that may occur in the Gorda Ridge Lease area, including hydrothermal vent organisms .................................55 BENTHOS THE STATE OF SCIENTIFIC INFORMATION RELATING TO THE BIOLOGY AND ECOLOGY 3F THE GOUDA RIDGE STUDY AREA, NORTZEAST PACIFIC OCEAN: INTRODUCTION Presently, only two published studies discuss the ecology of benthic animals on the Gorda Sidge. Fowler and Kulm (19701, in a predominantly geolgg isal study, used the presence of sublittor31 and planktsnic foraminiferans as an indication of uplift of tfie deep-sea fioor. Their resuits showed tiac sedinenta ana foraminiferans are depositea in the Zscanaba Trough, in the southern part of the Corda Ridge, by turbidity currents with a continental origin. They list 22 species of fararniniferans from the Gorda Rise (See Appendix 13. A more recent study collected geophysical, geological, and biological data from the Gorda Ridge, with particular emphasis on the northern part of the Ridge (Clague et al. 19843. Geological data suggest the presence of widespread low-temperature hydrothermal activity along the axf s of the northern two-thirds of the Corda 3idge. However, the relative age of these vents, their present activity and presence of sulfide deposits are currently unknown. The biological data, again with an emphasis on foraminiferans, indicate relatively high species diversity and high density , perhaps assoc iated with widespread hydrotheraal activity. -

METHANOGENS AS MODELS for LIFE on MARS. R. L. Mickol1, W. H. Waddell2, and T
Eighth International Conference on Mars (2014) 1005.pdf METHANOGENS AS MODELS FOR LIFE ON MARS. R. L. Mickol1, W. H. Waddell2, and T. A. Kral1,3, 1Arkansas Center for Space and Planetary Sciences, 202 Old Museum Building, University of Arkansas, Fayetteville, Arkansas, 72701, USA, [[email protected]], 2Dept. of Health, Human Performance, and Recreation, HPER 308, University of Arkansas, Fayetteville, Arkansas, 72701, USA, 3Dept. of Biological Sciences, SCEN 632, University of Arkansas, Fayetteville, Arkansas, 72701, USA. Introduction: The discovery of methane in the Sciences, University of Arkansas, Fayetteville, Arkan- martian atmosphere [1-4] has fueled the study of meth- sas. All four methanogen species were tested in these anogens as ideal candidates for life on Mars. Methano- experiments. Methanogens were grown in their respec- gens are chemoautotrophs from the domain Archaea. tive anaerobic growth media and placed into the cham- These microorganisms utilize hydrogen as an energy ber with a palladium catalyst box to remove residual source and carbon dioxide as a carbon source to pro- oxygen. The chamber was evacuated to a pre- duce methane. Methanogens can be considered ideal determined pressure and filled with 80:20 H2:CO2 gas. candidates for life on Mars because they are anaerobic, This procedure was repeated three times to ensure re- they do not require organic nutrients and are non- moval of the atmosphere. The chamber was then main- photosynthetic, indicating they could exist in sub- tained at the desired pressure (133-143 mbar, 67-72 surface environments. mbar, 33-38 mbar, 6-10 mbar, 7-20 mbar) for the dura- Our lab has studied methanogens as models for life tion of the experiments. -

The Resolved Mystery of Tardigrades
Journal of Investigative Genomics Mini review Open Access The resolved mystery of tardigrades Abstract Volume 4 Issue 2 - 2017 Tardigrades, also known as ‘Water bears’ or ‘Space Bears’ are famous for their ability to survive in extreme environmental conditions. With incredible ability to survive at a range Oisorjo Chakraborty, Pranab Roy Department of Biotechnology, Haldia Institute of Technology, of temperature between -272˚C to 150˚C, these small eight legged animals are considered India as one of the toughest organisms of the world. There are several records of survival of these animals under high pressure, UV exposure and even in space under cosmic radiation. For Correspondence: Pranab Roy, Department of Biotechnology, decades it remained a mystery how tardigrades can tolerate the extreme environmental Haldia Institute of Technology, India, Tel 9933037099, conditions. Afteryears of research, it was found that tardigrades dry out and turn their entire Email [email protected] body into a glass like structure at very low temperature, termed as “Bio-glass” to shut Oisorjo Chakraborty, Department of Biotechnology, Haldia down all their metabolic processes. Later by rehydrating, the creatures bring back life in Institute of Technology, India, Email [email protected] them. Finally researchers have identified the key protein in tardigrades which helps them to survive at extreme low temperature. The genome of several tardigrades species has been Received: April 21, 2017 | Published: May 19, 2017 sequenced like ‘Ramazzottius varieornatus’, ‘Hypsibius dujardini’. The sequenced genome provides evidences of expression of tolerance related genes and horizontal gene transfer in the animals helping them to survive in extreme environmental conditions with these key genes. -

Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities
Provided for non-commercial research and educational use only. Not for reproduction, distribution or commercial use. This chapter was originally published in the book Advances in Applied Microbiology, Vol 66, published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who know you, and providing a copy to your institution’s administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at: http://www.elsevier.com/locate/permissionusematerial From: Noha Youssef, Mostafa S. Elshahed, and Michael J. McInerney, Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities. In Allen I. Laskin, Sima Sariaslani, and Geoffrey M. Gadd, editors: Advances in Applied Microbiology, Vol 66, Burlington: Academic Press, 2009, pp. 141-251. ISBN: 978-0-12-374788-4 © Copyright 2009 Elsevier Inc. Academic Press. Author's personal copy CHAPTER 6 Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities Noha Youssef, Mostafa S. Elshahed, and Michael J. McInerney1 Contents I. Introduction 142 II. Factors Governing Oil Recovery 144 III. Microbial Ecology of Oil Reservoirs 147 A. Origins of microorganisms recovered from oil reservoirs 147 B. Microorganisms isolated from oil reservoirs 148 C. Culture-independent analysis of microbial communities in oil reservoirs 155 IV. -

Methanogens: Pushing the Boundaries of Biology
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Biochemistry -- Faculty Publications Biochemistry, Department of 12-14-2018 Methanogens: pushing the boundaries of biology Nicole R. Buan Follow this and additional works at: https://digitalcommons.unl.edu/biochemfacpub Part of the Biochemistry Commons, Biotechnology Commons, and the Other Biochemistry, Biophysics, and Structural Biology Commons This Article is brought to you for free and open access by the Biochemistry, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Biochemistry -- Faculty Publications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Emerging Topics in Life Sciences (2018) 2 629–646 https://doi.org/10.1042/ETLS20180031 Review Article Methanogens: pushing the boundaries of biology Nicole R. Buan Department of Biochemistry, University of Nebraska-Lincoln, 1901 Vine St., Lincoln, NE 68588-0664, U.S.A. Correspondence: Nicole R. Buan ([email protected]) Downloaded from https://portlandpress.com/emergtoplifesci/article-pdf/2/4/629/484198/etls-2018-0031c.pdf by University of Nebraska Libraries user on 11 February 2020 Methanogens are anaerobic archaea that grow by producing methane gas. These microbes and their exotic metabolism have inspired decades of microbial physiology research that continues to push the boundary of what we know about how microbes conserve energy to grow. The study of methanogens has helped to elucidate the thermodynamic and bioener- getics basis of life, contributed our understanding of evolution and biodiversity, and has garnered an appreciation for the societal utility of studying trophic interactions between environmental microbes, as methanogens are important in microbial conversion of biogenic carbon into methane, a high-energy fuel. -

Carbonate-Hosted Methanotrophy Represents an Unrecognized Methane Sink in the Deep Sea
ARTICLE Received 24 Mar 2014 | Accepted 28 Aug 2014 | Published 14 Oct 2014 DOI: 10.1038/ncomms6094 Carbonate-hosted methanotrophy represents an unrecognized methane sink in the deep sea Jeffrey J. Marlow1, Joshua A. Steele1, Wiebke Ziebis2, Andrew R. Thurber3, Lisa A. Levin4 & Victoria J. Orphan1 The atmospheric flux of methane from the oceans is largely mitigated through microbially mediated sulphate-coupled methane oxidation, resulting in the precipitation of authigenic carbonates. Deep-sea carbonates are common around active and palaeo-methane seepage, and have primarily been viewed as passive recorders of methane oxidation; their role as active and unique microbial habitats capable of continued methane consumption has not been examined. Here we show that seep-associated carbonates harbour active microbial communities, serving as dynamic methane sinks. Microbial aggregate abundance within the carbonate interior exceeds that of seep sediments, and molecular diversity surveys reveal methanotrophic communities within protolithic nodules and well-lithified carbonate pavements. Aggregations of microbial cells within the carbonate matrix actively oxidize 14 methane as indicated by stable isotope FISH–nanoSIMS experiments and CH4 radiotracer rate measurements. Carbonate-hosted methanotrophy extends the known ecological niche of these important methane consumers and represents a previously unrecognized methane sink that warrants consideration in global methane budgets. 1 Division of Geological and Planetary Sciences, California Institute of Technology, Pasadena, California 91125, USA. 2 Department of Biological Science, University of Southern California, Los Angeles, California 90089, USA. 3 College of Earth, Ocean, and Atmospheric Sciences, Oregon State University, Corvallis, Oregon 97331, USA. 4 Center for Marine Biodiversity and Conservation, Integrative Oceanography Division, Scripps Institution of Oceanography, University of California–San Diego, La Jolla, California 92037, USA. -

Microbial Processes and Natural Gas Accumulations Barry Jay Katz*
The Open Geology Journal, 2011, 5, 75-83 75 Open Access Microbial Processes and Natural Gas Accumulations Barry Jay Katz* Chevron Energy Technology Company, 1500 Louisiana Street, Houston, TX 77002, USA Abstract: Microbial processes are responsible for the formation, alteration, and destruction of some natural gas accumulations. Individual microbial gas accumulations may be significant (> 1 Tcf; trillion cubic feet or 28.3 billion cubic meters) and collectively account for more than 20% of the global gas resource-base, dominating in some individual basins. Often cited resource estimates do not effectively account for the contribution of microbial gas to mixed biogenic- thermogenic gases nor do these estimates fully account for secondary microbial gas. Microbial gas accumulations form through multiple stages, initiating with the breakdown of large macromolecules into smaller components that the methanogens can effectively utilize as an energy source. There are two primary methanogenic pathways, acetate fermentation and CO2 reduction, both of which may utilize sedimentary organic matter or preexisting oil accumulations as a source. Microbial gas is typically considered dry (i.e., depleted in C2+ components), but there is clear evidence that ethane and possibly propane may also form through microbial processes. Numerous authors have provided guidance on the conditions that favor primary microbial gas formation and accumulation including: an anoxic setting, high rates of sedimentation, formation of early traps, low temperatures, favorable migration history, and limited availability of sulfate. Scenarios conducive with secondary microbial gas accumulations have also been discussed. These include the presence of a significant oil pool that can be biodegraded or a coal or organic-rich shale that has achieved thermal maturity levels approaching or beyond the top of the oil-window, which have been uplifted, fractured, and invaded by meteoric water to reintroduce a microbial population. -
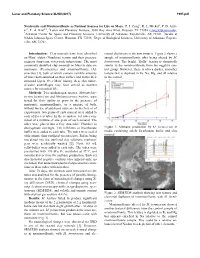
Nontronite and Montmorillonite As Nutrient Sources for Life on Mars
Lunar and Planetary Science XLVIII (2017) 1997.pdf Nontronite and Montmorillonite as Nutrient Sources for Life on Mars. P. I. Craig1, R. L. Mickol2, P. D. Arch- er3, T. A. Kral2,4, 1Lunar and Planetary Institute, 3600 Bay Area Blvd, Houston TX 77058 ([email protected]), 2Arkansas Center for Space and Planetary Sciences, University of Arkansas, Fayetteville, AR 72701, 3Jacobs at NASA Johnson Space Center, Houston, TX 77058, 4Dept. of Biological Sciences, University of Arkansas, Fayette- ville, AR, 72701. Introduction: Clay minerals have been identified mental depletions in the new textures. Figure 2 shows a on Mars’ oldest (Noachian) terrain and their presence sample of montmorillonite after being altered by M. suggests long-term water-rock interactions. The most formicicum. The bright, “fluffy” texture is chemically commonly identified clay minerals on Mars to date are similar to the montmorillonite from the negative con- nontronite (Fe-smectite) and montmorillonite (Al- trol group. However, there is also a darker, smoother smectite) [1], both of which contain variable amounts texture that is depleted in Fe, Na, Mg, and Al relative of water both adsorbed on their surface and within their to the control. structural layers. Over Mars’ history, these clay miner- al-water assemblages may have served as nutrient sources for microbial life. Methods: Two methanogen species, Methanobac- terium formicicum and Methanosarcina barkeri, were tested for their ability to grow in the presence of nontronite, montmorillonite, or a mixture of both, without the use of additional nutrients. In the first set of experiments, two grams of each mineral were added to each of five test tubes. -

Methanogenic Microorganisms in Industrial Wastewater Anaerobic Treatment
processes Review Methanogenic Microorganisms in Industrial Wastewater Anaerobic Treatment Monika Vítˇezová 1 , Anna Kohoutová 1, Tomáš Vítˇez 1,2,* , Nikola Hanišáková 1 and Ivan Kushkevych 1,* 1 Department of Experimental Biology, Faculty of Science, Masaryk University, 62500 Brno, Czech Republic; [email protected] (M.V.); [email protected] (A.K.); [email protected] (N.H.) 2 Department of Agricultural, Food and Environmental Engineering, Faculty of AgriSciences, Mendel University, 61300 Brno, Czech Republic * Correspondence: [email protected] (T.V.); [email protected] (I.K.); Tel.: +420-549-49-7177 (T.V.); +420-549-49-5315 (I.K.) Received: 31 October 2020; Accepted: 24 November 2020; Published: 26 November 2020 Abstract: Over the past decades, anaerobic biotechnology is commonly used for treating high-strength wastewaters from different industries. This biotechnology depends on interactions and co-operation between microorganisms in the anaerobic environment where many pollutants’ transformation to energy-rich biogas occurs. Properties of wastewater vary across industries and significantly affect microbiome composition in the anaerobic reactor. Methanogenic archaea play a crucial role during anaerobic wastewater treatment. The most abundant acetoclastic methanogens in the anaerobic reactors for industrial wastewater treatment are Methanosarcina sp. and Methanotrix sp. Hydrogenotrophic representatives of methanogens presented in the anaerobic reactors are characterized by a wide species diversity. Methanoculleus