Weighted Likelihood Inference of Genomic Autozygosity Patterns in Dense Genotype Data Alexandra Blant1†, Michelle Kwong1†, Zachary A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
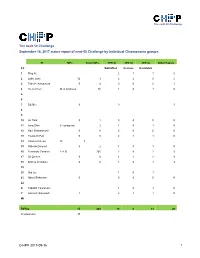
Next-MP50 Status Report
The neXt-50 Challenge The neXt-50 Challenge September 16, 2017 status report of next-50 Challenge by individual Chromosome groups. PI MPs Silver MPs JPR SI JPR SI JPR SI Other Papers Ch Submitted In press In revision 1 Ping Xu 2 1 1 0 2 Lydie Lane 12 3 2 2 0 2 3 Takeshi Kawamura 0 4 0 0 0 1 4 Yu-Ju Chen 26 in progress 86 1 0 1 0 5 6 7 Ed Nice 0 0 1 8 9 10 Jin Park 0 1 0 0 0 0 11 Jong Shin 2 in progress 2 1 0 1 0 12 Ravi Sirdeshmukh 0 0 0 0 0 0 13 Young-Ki Paik 0 5 2 1 1 0 14 CharLes Pineau 12 3 15 GiLberto Domont 0 2 1 0 1 0 16 Fernando CorraLes 2 (+ 9) 165 2 0 2 5 17 GiL Omenn 0 0 3 1 2 8 18 Andrey Archakov 0 0 1 0 1 3 19 20 Siqi Liu 1 0 1 21 Albert Sickmann 0 0 0 0 0 22 X Tadashi Yamamoto 1 0 1 0 Y Hosseini SaLekdeh 1 2 1 1 0 Mt TOTAL 15 268 19 6 13 20 (in progress) 37 C-HPP 2017-09-16 1 The neXt-50 Challenge Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). -

Persistence of Breakage in Specific Chromosome Bands 6 Years After Acute Exposure to Oil
RESEARCH ARTICLE Persistence of Breakage in Specific Chromosome Bands 6 Years after Acute Exposure to Oil Alexandra Francés1, Kristin Hildur1, Joan Albert Barberà2,3, Gema Rodríguez-Trigo2,4, Jan-Paul Zock5,6,7, Jesús Giraldo8, Gemma Monyarch1, Emma Rodriguez-Rodriguez9, Fernanda de Castro Reis1, Ana Souto9, Federico P. Gómez2,3, Francisco Pozo- Rodríguez2,10, Cristina Templado1, Carme Fuster1* 1 Unitat de Biologia CelÁlular i Genètica Mèdica, Facultat de Medicina, Universitat Autònoma de Barcelona (UAB), Bellaterra, Spain, 2 CIBER Enfermedades Respiratorias (CIBERES), Bunyola, Mallorca, Spain, a11111 3 Departament de Medicina Respiratòria, Hospital Clínic-Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain, 4 Departamento de Medicina Respiratoria, Hospital Clínico San Carlos, Madrid, Spain, 5 Centre de Recerca en Epidemiologia Ambiental (CREAL), Barcelona, Spain, 6 Institut Recerca Medica, Hospital del Mar, Barcelona, Spain, 7 CIBER Epidemiologia i Salut Pública (CIBERESP), Barcelona, Spain, 8 Institut de Neurociències and Unitat de Bioestadística, Facultat de Medicina, UAB, Bellaterra, Spain, 9 Departamento de Medicina Respiratoria, Complexo Hospitalario Universitario A Coruña, A Coruña, Spain, 10 Departamento de Medicina Respiratoria, Unidad Epidemiologia Clínica, Hospital 12 de Octubre, Madrid, Spain OPEN ACCESS * [email protected] Citation: Francés A, Hildur K, Barberà JA, Rodríguez-Trigo G, Zock J-P, Giraldo J, et al. (2016) Persistence of Breakage in Specific Chromosome Bands 6 Years after Acute Exposure to Oil. PLoS Abstract ONE 11(8): e0159404. doi:10.1371/journal. pone.0159404 Editor: Roberto Amendola, ENEA, ITALY Background Received: December 21, 2015 The identification of breakpoints involved in chromosomal damage could help to detect Accepted: July 2, 2016 genes involved in genetic disorders, most notably cancer. -
A Resource for Exploring the Understudied Human Kinome for Research and Therapeutic
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.02.022277; this version posted March 11, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A resource for exploring the understudied human kinome for research and therapeutic opportunities Nienke Moret1,2,*, Changchang Liu1,2,*, Benjamin M. Gyori2, John A. Bachman,2, Albert Steppi2, Clemens Hug2, Rahil Taujale3, Liang-Chin Huang3, Matthew E. Berginski1,4,5, Shawn M. Gomez1,4,5, Natarajan Kannan,1,3 and Peter K. Sorger1,2,† *These authors contributed equally † Corresponding author 1The NIH Understudied Kinome Consortium 2Laboratory of Systems Pharmacology, Department of Systems Biology, Harvard Program in Therapeutic Science, Harvard Medical School, Boston, Massachusetts 02115, USA 3 Institute of Bioinformatics, University of Georgia, Athens, GA, 30602 USA 4 Department of Pharmacology, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA 5 Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University, Chapel Hill, NC 27599, USA † Peter Sorger Warren Alpert 432 200 Longwood Avenue Harvard Medical School, Boston MA 02115 [email protected] cc: [email protected] 617-432-6901 ORCID Numbers Peter K. Sorger 0000-0002-3364-1838 Nienke Moret 0000-0001-6038-6863 Changchang Liu 0000-0003-4594-4577 Benjamin M. Gyori 0000-0001-9439-5346 John A. Bachman 0000-0001-6095-2466 Albert Steppi 0000-0001-5871-6245 Shawn M. -

University of Dundee MASTER of SCIENCE a Pilot Study- Identify
University of Dundee MASTER OF SCIENCE A Pilot Study- Identify Genetic Variants for Diabetic Cataract Using GoDARTS Dataset Zhang, Kaida Award date: 2016 Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 11. Oct. 2021 A Pilot Study- Identify Genetic Variants for Diabetic Cataract Using GoDARTS Dataset Kaida Zhang A dissertation submitted for the degree of Master of Science by Research from the University of Dundee School of Medicine University of Dundee September 2016 Abstract Background: Diabetic cataract is one of the major eye complications of diabetes. It was reported that cataract occurs two to five times more frequently in patients with diabetes compared with those with no diabetes. The purpose of this study was to identify genetic contributors of diabetic cataract based on a genome-wide association approach using a well-defined Scottish diabetic cohort. -

Development of a Cellular Model for C9ORF72-Related Amyotrophic Lateral Sclerosis
Development of a Cellular Model for C9ORF72-related Amyotrophic Lateral Sclerosis By Matthew John Stopford Sheffield Institute for Translational Neuroscience University of Sheffield Submitted for the degree of Doctor of Philosophy (PhD) May 2016 Acknowledgements Firstly, I would like to acknowledge and thank my supervisors Dr. Janine Kirby, Prof. Dame Pamela Shaw, and Dr. Adrian Higginbottom for their continued patience and support throughout my PhD. I am incredibly grateful to Janine for providing both academic and personal support during my PhD. I am thankful to Pam for her guidance, but also for pushing me to develop personally and as a scientist. Also, I am very thankful to Adrian, who has been a great mentor, helping me to develop my technical skills, my ability to think critically, and has also been incredibly patient when I have found the PhD hardest. I would also like to thank the Sheffield Hospitals Charity for funding my PhD project. There are several other individuals who have been incredibly helpful during my time in SITraN as well, who deserve acknowledgment. Dr. Guillaume Hautbergue has been optimistic and inspirational, and has taught me many techniques in the lab. Dr. Matthew Walsh has taught me several biochemical techniques, and has been incredibly supportive personally. Dr. Jonathan Cooper-Knock has been very helpful in explaining protocols, array analysis and generally supporting my work. Also, I would like to thank Dr. Paul Heath and Mrs. Catherine Gelsthorpe for their technical assistance and support with the transcriptomic work. This PhD has genuinely been the greatest challenge of my life, and without the support of my parents and family, the Sims family, my friends and other PhD students, I would have failed. -

MAP3K19 Is a Novel Regulator of TGF-Β Signaling That Impacts Bleomycin-Induced Lung Injury and Pulmonary Fibrosis
RESEARCH ARTICLE MAP3K19 Is a Novel Regulator of TGF-β Signaling That Impacts Bleomycin-Induced Lung Injury and Pulmonary Fibrosis Stefen A. Boehme1*, Karin Franz-Bacon2, Danielle N. DiTirro1¤, Tai Wei Ly1, Kevin B. Bacon1 1 AxikinPharmaceuticals, Inc., San Diego, California, United States of America, 2 DNA Consulting, Inc., San Diego, California, United States of America a11111 ¤ Current address: Department of Biology, Brandeis University, Waltham, MA, United States of America * [email protected] Abstract Idiopathic pulmonary fibrosis (IPF) is a progressive, debilitating disease for which two medi- OPEN ACCESS cations, pirfenidone and nintedanib, have only recently been approved for treatment. The Citation: Boehme SA, Franz-Bacon K, DiTirro DN, cytokine TGF-β has been shown to be a central mediator in the disease process. We inves- Ly TW, Bacon KB (2016) MAP3K19 Is a Novel tigated the role of a novel kinase, MAP3K19, upregulated in IPF tissue, in TGF-β-induced β Regulator of TGF- Signaling That Impacts signal transduction and in bleomycin-induced pulmonary fibrosis. MAP3K19 has a very lim- Bleomycin-Induced Lung Injury and Pulmonary Fibrosis. PLoS ONE 11(5): e0154874. doi:10.1371/ ited tissue expression, restricted primarily to the lungs and trachea. In pulmonary tissue, journal.pone.0154874 expression was predominantly localized to alveolar and interstitial macrophages, bronchial Editor: Wei Shi, Children's Hospital Los Angeles, epithelial cells and type II pneumocytes of the epithelium. MAP3K19 was also found to be UNITED STATES overexpressed in bronchoalveolar lavage macrophages from IPF patients compared to nor- Received: November 19, 2015 mal patients. Treatment of A549 or THP-1 cells with either MAP3K19 siRNA or a highly potent and specific inhibitor reduced phospho-Smad2 & 3 nuclear translocation following Accepted: April 20, 2016 TGF-β stimulation.