Inflammation and Aging of Hematopoietic Stem Cells in Their
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Influence of Infection and Inflammation on Biomarkers of Nutritional Status
A2.4 INFLUENCE OF INFECTION AND INFLAMMATION ON BIOMARKERS OF NUTRITIONAL STATUS A2.4 Influence of infection and inflammation on biomarkers of nutritional status with an emphasis on vitamin A and iron David I. Thurnham1 and George P. McCabe2 1 Northern Ireland Centre for Food and Health, University of Ulster, Coleraine, United Kingdom of Great Britain and Northern Ireland 2 Statistics Department, Purdue University, West Lafayette, Indiana, United States of America Corresponding author: David I. Thurnham; [email protected] Suggested citation: Thurnham DI, McCabe GP. Influence of infection and inflammation on biomarkers of nutritional status with an emphasis on vitamin A and iron. In: World Health Organization. Report: Priorities in the assessment of vitamin A and iron status in populations, Panama City, Panama, 15–17 September 2010. Geneva, World Health Organization, 2012. Abstract n Many plasma nutrients are influenced by infection or tissue damage. These effects may be passive and the result of changes in blood volume and capillary permeability. They may also be the direct effect of metabolic alterations that depress or increase the concentration of a nutrient or metabolite in the plasma. Where the nutrient or metabolite is a nutritional biomarker as in the case of plasma retinol, a depression in retinol concentrations will result in an overestimate of vitamin A deficiency. In contrast, where the biomarker is increased due to infection as in the case of plasma ferritin concentrations, inflammation will result in an underestimate of iron deficiency. Infection and tissue damage can be recognized by their clinical effects on the body but, unfortunately, subclinical infection or inflammation can only be recognized by measur- ing inflammation biomarkers in the blood. -

The Gut Microbiota and Inflammation
International Journal of Environmental Research and Public Health Review The Gut Microbiota and Inflammation: An Overview 1, 2 1, 1, , Zahraa Al Bander *, Marloes Dekker Nitert , Aya Mousa y and Negar Naderpoor * y 1 Monash Centre for Health Research and Implementation, School of Public Health and Preventive Medicine, Monash University, Melbourne 3168, Australia; [email protected] 2 School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane 4072, Australia; [email protected] * Correspondence: [email protected] (Z.A.B.); [email protected] (N.N.); Tel.: +61-38-572-2896 (N.N.) These authors contributed equally to this work. y Received: 10 September 2020; Accepted: 15 October 2020; Published: 19 October 2020 Abstract: The gut microbiota encompasses a diverse community of bacteria that carry out various functions influencing the overall health of the host. These comprise nutrient metabolism, immune system regulation and natural defence against infection. The presence of certain bacteria is associated with inflammatory molecules that may bring about inflammation in various body tissues. Inflammation underlies many chronic multisystem conditions including obesity, atherosclerosis, type 2 diabetes mellitus and inflammatory bowel disease. Inflammation may be triggered by structural components of the bacteria which can result in a cascade of inflammatory pathways involving interleukins and other cytokines. Similarly, by-products of metabolic processes in bacteria, including some short-chain fatty acids, can play a role in inhibiting inflammatory processes. In this review, we aimed to provide an overview of the relationship between the gut microbiota and inflammatory molecules and to highlight relevant knowledge gaps in this field. -

Mixing Old and Young: Enhancing Rejuvenation and Accelerating Aging
Mixing old and young: enhancing rejuvenation and accelerating aging Ashley Lau, … , James L. Kirkland, Stefan G. Tullius J Clin Invest. 2019;129(1):4-11. https://doi.org/10.1172/JCI123946. Review Donor age and recipient age are factors that influence transplantation outcomes. Aside from age-associated differences in intrinsic graft function and alloimmune responses, the ability of young and old cells to exert either rejuvenating or aging effects extrinsically may also apply to the transplantation of hematopoietic stem cells or solid organ transplants. While the potential for rejuvenation mediated by the transfer of youthful cells is currently being explored for therapeutic applications, aspects that relate to accelerating aging are no less clinically significant. Those effects may be particularly relevant in transplantation with an age discrepancy between donor and recipient. Here, we review recent advances in understanding the mechanisms by which young and old cells modify their environments to promote rejuvenation- or aging-associated phenotypes. We discuss their relevance to clinical transplantation and highlight potential opportunities for therapeutic intervention. Find the latest version: https://jci.me/123946/pdf REVIEW The Journal of Clinical Investigation Mixing old and young: enhancing rejuvenation and accelerating aging Ashley Lau,1 Brian K. Kennedy,2,3,4,5 James L. Kirkland,6 and Stefan G. Tullius1 1Division of Transplant Surgery, Department of Surgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA. 2Departments of Biochemistry and Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore. 3Singapore Institute for Clinical Sciences, Singapore. 4Agency for Science, Technology and Research (A*STAR), Singapore. -

SENS-Research-Foundation-2019
by the year 2050, cardiovascular an estimated 25-30 the american 85 percent of adults disease years and older age 85 or older remains the most population will suffer from common cause of 2 1 2 dementia. death in older adults. triple. THE CLOCK IS TICKING. By 2030, annual direct The estimated cost of medical costs associated dementia worldwide was 62% of Americans with cardiovascular $818 billion diseases in the united over age 65 have in 2015 and is states are expected to more than one expected to grow to rise to more than chronic condition.1 3 $2 trillion $818 billion. by 2030.1 References: (1) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5732407/, (2) https://www.who.int/ageing/publications/global_health.pdf, (3) https://www.cdcfoundation.org/pr/2015/heart-disease-and-stroke-cost-america-nearly-1-billion-day-medical-costs-lost-productivity sens research foundation board of directors Barbara Logan Kevin Perrott Bill Liao Chairperson Treasurer Secretary Michael Boocher Kevin Dewalt James O’Neill Jonathan Cain Michael Kope Frank Schuler 02 CONTENTS 2019 Annual Report 04 Letter From The CEO 06 Outreach & Fundraising 08 Finances 09 Donors erin ashford photography 14 Education 26 Investments 20 Conferences & Events 30 Research Advisory Board 23 Speaking Engagements 31 10 Years Of Research 24 Alliance 32 MitoSENS 34 LysoSENS 35 Extramural Research 38 Publications 39 Ways to Donate cover Photo (c) Mikhail Leonov - stock.adobe.com special 10th anniversary edition 03 FROM THE CEO It’s early 2009, and it’s very late at night. Aubrey, Jeff, Sarah, Kevin, and Mike are sitting around a large table covered in papers and half-empty food containers. -

Innate Immunity and Inflammation
ISBTc ‐ Primer on Tumor Immunology and Biological Therapy of Cancer InnateInnate ImmunityImmunity andand InflammationInflammation WillemWillem Overwijk,Overwijk, Ph.D.Ph.D. MDMD AndersonAnderson CancerCancer CenterCenter CenterCenter forfor CancerCancer ImmunologyImmunology ResearchResearch Houston,Houston, TXTX www.allthingsbeautiful.com InnateInnate ImmunityImmunity andand InflammationInflammation • Definitions • Cells and Molecules • Innate Immunity and Inflammation in Cancer • Bad Inflammation • Good Inflammation • Therapeutic Implications InnateInnate ImmunityImmunity andand InflammationInflammation • Definitions • Cells and Molecules • Innate Immunity and Inflammation in Cancer • Bad Inflammation • Good Inflammation • Therapeutic Implications • Innate Immunity: Immunity that is naturally present and is not due to prior sensitization to an antigen; generally nonspecific. It is in contrast to acquired/adaptive immunity. Adapted from Merriam‐Webster Medical Dictionary • Innate Immunity: Immunity that is naturally present and is not due to prior sensitization to an antigen; generally nonspecific. It is in contrast to acquired/adaptive immunity. • Inflammation: a local response to tissue injury – Rubor (redness) – Calor (heat) – Dolor (pain) – Tumor (swelling) Adapted from Merriam‐Webster Medical Dictionary ““InnateInnate ImmunityImmunity”” andand ““InflammationInflammation”” areare vaguevague termsterms •• SpecificSpecific cellcell typestypes andand moleculesmolecules orchestrateorchestrate specificspecific typestypes ofof inflammationinflammation -

Read Our New Annual Report
The seeds of a concept. The roots of an idea. The potential of a world free of age-related disease. Photo: Sherry Loeser Photography SENS Research Foundation Board of Directors Barbara Logan, Chair Bill Liao, Secretary Kevin Perrott, Treasurer Michael Boocher Jonathan Cain Kevin Dewalt Michael Kope Jim O’Neill Frank Schüler Sherry Loeser Photography 2 Contents CEO Letter (Jim O’Neill) 4 Finances 5 Donors 6 - 7 Fundraising & Conferences 8 - 9 Around the World with Aubrey de Grey 10 Outreach 11 Founding CEO Tribute & Underdog Pharmaceuticals 12 - 13 Investments 14 Welcome New Team Members 15 Education 16 - 17 Publications & Research Advisory Board 18 Research Summaries 19 - 22 Ways to Donate 23 The SRF Team Front row: Anne Corwin (Engineer/Editor), Amutha Boominathan (MitoSENS Group Lead), Alexandra Stolzing (VP of Research), Aubrey de Grey (Chief Science Officer), Jim O’Neill (CEO), Bhavna Dixit (Research Associate). Center row: Caitlin Lewis (Research Associate), Lisa Fabiny-Kiser (VP of Operations), Gary Abramson (Graphics), Maria Entraigues-Abramson (Global Outreach Coordinator), Jessica Lubke (Administrative Assistant). Back row: Tesfahun Dessale Admasu (Research Fellow), Amit Sharma (ImmunoSENS Group Lead), Michael Rae (Science Writer), Kelly Protzman (Executive Assistant). Not Pictured: Greg Chin (Director, SRF Education), Ben Zealley (Website/Research Assistant/ Deputy Editor) Photo: Sherry Loeser Photography, 2019 3 From the CEO At our 2013 conference at Queens College, Cambridge, I closed my talk by saying, “We should not rest until we make aging an absurdity.” We are now in a very different place. After a lot of patient explanation, publication of scientific results, conferences, and time, our community persuaded enough scientists of the feasibility of the damage repair approach to move SENS and SENS Research Foundation from the fringes of scientific respectability to the vanguard of a mainstream community of scientists developing medical therapies to tackle human aging. -

Age-Dependent Deterioration of Nuclear Pore Assembly in Mitotic
RESEARCH ARTICLE Age-dependent deterioration of nuclear pore assembly in mitotic cells decreases transport dynamics Irina L Rempel1, Matthew M Crane2, David J Thaller3, Ankur Mishra4, Daniel PM Jansen1, Georges Janssens1, Petra Popken1, Arman Aks¸it1, Matt Kaeberlein2, Erik van der Giessen4, Anton Steen1, Patrick R Onck4, C Patrick Lusk3, Liesbeth M Veenhoff1* 1European Research Institute for the Biology of Ageing (ERIBA), University of Groningen, University Medical Center Groningen, Groningen, Netherlands; 2Department of Pathology, University of Washington, Seattle, United States; 3Department of Cell Biology, Yale School of Medicine, New Haven, United States; 4Zernike Institute for Advanced Materials, University of Groningen, Groningen, Netherlands Abstract Nuclear transport is facilitated by the Nuclear Pore Complex (NPC) and is essential for life in eukaryotes. The NPC is a long-lived and exceptionally large structure. We asked whether NPC quality control is compromised in aging mitotic cells. Our images of single yeast cells during aging, show that the abundance of several NPC components and NPC assembly factors decreases. Additionally, the single-cell life histories reveal that cells that better maintain those components are longer lived. The presence of herniations at the nuclear envelope of aged cells suggests that misassembled NPCs are accumulated in aged cells. Aged cells show decreased dynamics of transcription factor shuttling and increased nuclear compartmentalization. These functional changes are likely caused by the presence -

Effect of Exercise Intensity on Cell-Mediated Immunity
sports Perspective Effect of Exercise Intensity on Cell-Mediated Immunity Katsuhiko Suzuki 1,* and Harumi Hayashida 2 1 Faculty of Sport Sciences, Waseda University, 2-579-15 Mikajima, Tokorozawa 359-1192, Japan 2 Faculty of Culture and Sport Policy, Toin University of Yokohama, 1614 Kurogane-cho, Aoba-ku, Yokohama 225-8503, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-4-2947-6898 Abstract: Moderate-intensity exercise is considered to enhance immune function and to be useful for preventing acute upper respiratory infections and similar conditions. Many people practice low- intensity short-duration exercise with the expectation of a beneficial effect on immunocompetency. However, it is difficult to affirm the existence of definite evidence of such a benefit. In this article, we discuss the effects of low-intensity short-duration exercise on cell-mediated immunity, and contrast them to the effects of high-intensity and long-duration exercise. Whereas high-intensity exercise induces inflammation and reduces cell-mediated immune system function, low-intensity exercise does not appear to have a large effect on either inflammation or cell-mediated immune function. Low-intensity exercises such as walking and yoga, which are helpful to relieve stress, cannot be considered as harmful to the immune system. Although yoga was shown to impose fewer restrictions on breathing and physical strain, the evidence that yoga enhances cell-mediated immunity remains insufficient. Therefore, further studies are needed to examine the exercise mode that may be most effective for improvement of immune functions. Keywords: exercise; walking; yoga; cellular immune system; cytokines; inflammation 1. -

An EANM Procedural Guideline
European Journal of Nuclear Medicine and Molecular Imaging https://doi.org/10.1007/s00259-018-4052-x GUIDELINES Clinical indications, image acquisition and data interpretation for white blood cells and anti-granulocyte monoclonal antibody scintigraphy: an EANM procedural guideline A. Signore1 & F. Jamar2 & O. Israel3 & J. Buscombe4 & J. Martin-Comin5 & E. Lazzeri6 Received: 27 April 2018 /Accepted: 6 May 2018 # The Author(s) 2018 Abstract Introduction Radiolabelled autologous white blood cells (WBC) scintigraphy is being standardized all over the world to ensure high quality, specificity and reproducibility. Similarly, in many European countries radiolabelled anti-granulocyte antibodies (anti-G-mAb) are used instead of WBC with high diagnostic accuracy. The EANM Inflammation & Infection Committee is deeply involved in this process of standardization as a primary goal of the group. Aim The main aim of this guideline is to support and promote good clinical practice despite the complex environment of a national health care system with its ethical, economic and legal aspects that must also be taken into consideration. Method After the standardization of the WBC labelling procedure (already published), a group of experts from the EANM Infection & Inflammation Committee developed and validated these guidelines based on published evidences. Results Here we describe image acquisition protocols, image display procedures and image analyses as well as image interpre- tation criteria for the use of radiolabelled WBC and monoclonal antigranulocyte antibodies. Clinical application for WBC and anti-G-mAb scintigraphy is also described. Conclusions These guidelines should be applied by all nuclear medicine centers in favor of a highly reproducible standardized practice. Keywords Infection . -

Transhumanism, Metaphysics, and the Posthuman God
Journal of Medicine and Philosophy, 35: 700–720, 2010 doi:10.1093/jmp/jhq047 Advance Access publication on November 18, 2010 Transhumanism, Metaphysics, and the Posthuman God JEFFREY P. BISHOP* Saint Louis University, St. Louis, Missouri, USA *Address correspondence to: Jeffrey P. Bishop, MD, PhD, Albert Gnaegi Center for Health Care Ethics, Saint Louis University, 3545 Lafayette Avenure, Suite 527, St. Louis, MO 63104, USA. E-mail: [email protected] After describing Heidegger’s critique of metaphysics as ontotheol- ogy, I unpack the metaphysical assumptions of several transhu- manist philosophers. I claim that they deploy an ontology of power and that they also deploy a kind of theology, as Heidegger meant it. I also describe the way in which this metaphysics begets its own politics and ethics. In order to transcend the human condition, they must transgress the human. Keywords: Heidegger, metaphysics, ontology, ontotheology, technology, theology, transhumanism Everywhere we remain unfree and chained to technology, whether we passionately affirm or deny it. —Martin Heidegger I. INTRODUCTION Transhumanism is an intellectual and cultural movement, whose proponents declare themselves to be heirs of humanism and Enlightenment philosophy (Bostrom, 2005a, 203). Nick Bostrom defines transhumanism as: 1) The intellectual and cultural movement that affirms the possibility and desirability of fundamentally improving the human condition through applied reason, especially by developing and making widely available technologies to eliminate aging and to greatly enhance human intellectual, physical, and psychological capacities. 2) The study of the ramifications, promises, and potential dangers of technologies that will enable us to overcome fundamental human limitations, and the related study of the ethical matters involved in developing and using such technologies (Bostrom, 2003, 2). -
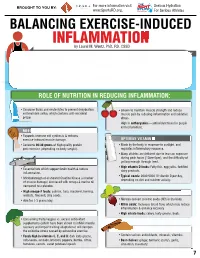
INFLAMMATION MICRONUTRIENTS SOURCES FUNCTION by Laurel M
For more information visit Serious Hydration For more information visit Serious Hydration BROUGHT TO YOU BY: BROUGHT TO YOU BY: For more information visit Serious Hydration www.SportsRD.org. For Serious Athletes www.SportsRD.org. For Serious Athletes www.SportsRD.org. NASA DEVELOPED For Serious Athletes NUTRITIONAL SUPPORT FOR INJURY BALANCING EXERCISE-INDUCED RECOVERY AND RETURN-TO-PLAY INFLAMMATION MICRONUTRIENTS SOURCES FUNCTION by Laurel M. Wentz, PhD, RD, CSSD Vitamin C Citrus fruit, red and green peppers, Antioxidant, wound healing, tissue repair, EXERCISE-INDUCED INFLAMMATION IS THE BODY’S RESPONSE TO INJURY DUE TO INTENSE PHYSICAL ACTIVITY cantaloupe immune function • The immune system response causes redness, swelling, pain. Vitamin A Sweet potato, spinach, carrots, Cell growth and development, • Acute inflammation is a normal response to high-intensity exercise, but prolonged (chronic) tomatoes immune function inflammation is a sustained response that affects the entire body. Vitamin D Sun exposure, oily fish, Promotes calcium absorption and bone health • Prolonged Inflammation: dairy products, fortified foods 1. Causes fatigue, muscle damage and soreness. 2. Limits muscle growth and training progression and increases muscle loss. Calcium Low-fat milk, fortified non-dairy milk, Supports skeletal structure and function 3. Modulating prolonged inflammation may enhance recovery & reduce soreness. low-fat Greek yogurt, cheese, broccoli, kale, fortified orange juice Magnesium Almonds, sesame and sunflower Nucleic acid and protein -

A Role of Inflammation and Immunity in Essential Hypertension—Modeled and Analyzed Using Petri Nets
International Journal of Molecular Sciences Article A Role of Inflammation and Immunity in Essential Hypertension—Modeled and Analyzed Using Petri Nets Dorota Formanowicz 1 , Agnieszka Rybarczyk 2,3 , Marcin Radom 2,3 and Piotr Formanowicz 2,3,* 1 Department of Clinical Biochemistry and Laboratory Medicine, Poznan University of Medical Sciences, 60-806 Poznan, Poland; [email protected] 2 Institute of Computing Science, Poznan University of Technology, 60-965 Poznan, Poland; [email protected] (A.R.); [email protected] (M.R.) 3 Institute of Bioorganic Chemistry, Polish Academy of Sciences, 61-704 Poznan, Poland * Correspondence: [email protected] Received: 15 April 2020; Accepted: 5 May 2020; Published: 9 May 2020 Abstract: Recent studies have shown that the innate and adaptive immune system, together with low-grade inflammation, may play an important role in essential hypertension. In this work, to verify the importance of selected factors for the development of essential hypertension, we created a Petri net-based model and analyzed it. The analysis was based mainly on t-invariants, knockouts of selected fragments of the net and its simulations. The blockade of the renin-angiotensin (RAA) system revealed that the most significant effect on the emergence of essential hypertension has RAA activation. This blockade affects: (1) the formation of angiotensin II, (2) inflammatory process (by influencing C-reactive protein (CRP)), (3) the initiation of blood coagulation, (4) bradykinin generation via the kallikrein-kinin system, (5) activation of lymphocytes in hypertension, (6) the participation of TNF alpha in the activation of the acute phase response, and (7) activation of NADPH oxidase—a key enzyme of oxidative stress.