129Th Annual Meeting Monday, October 4, 2004 Abstracts
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

155-L Page CHAPTER XII the 123D Division
N SI 55 U.S. Army Forces Far East. Military History Section. Record of Opera- tions Against Soviet Russia on Northern and Western Fronts of Manchuria, and in Northern Korea (August 1945). Japanese monograph no. 155. 1950. Distributed by the Office of the Chief of Military History, Departmen t of the Army. SAI ACCESS NO r77 A N Ju:i 2 ZQO ACCESSION NO -~iili~asi~w(i~I1786 7 '' ~r9 r k-':: ~a~ -jgy "'; :r' i.i 'i JAPANESE MONOGRAPH NO. 155 Ate.1 +++"-.i ti.,<a.. .. , i4,e NO su w..w..v- RECORD OF OPERATIONS AGAINST SO VIET RUSSIA ON NORTHERN AND WESTEF:N FRONTS OF MANCHURIA, AND IN NORTHERN KOREA (AUGUST 1945) PREPARED BY- MILITARY HISTORY SECTION HEADQUARTERS, ARMY FORCES FAR EAST DISTRIBUTED BY OFFICE OF THE CHIEF OF MILITARY HISTORY DEPARTMENT OF THE ARMY This monograph may not be reproduced without the permission of the Office Chief of Militsry History Monograph No. 155 Editor's Preface This is the last in a series of three monographs' covering Japanese military activities in Manchuria from January 1943 to the end of WVorld War II hostilities, prepared by former commanders and staff officers of the Kwantung Army. The first (No. 138) deals with Kwantung Arm's wartime vigil throughout Manchuria in prepa- ration for operations. The second (No. 154) deals with actual military operations against Soviet forces on the eastern front. This monograph, No. 155, covers operations in the northern and western parts of Manchuria and also in northern Korea. Like No. 154, this monograph is actually a collection of closely related sub-monographs, each a separate--but by no means complete--study in itself. -

Becoming the World's Most Competitive Restaurant Service Company
BECOMING THE WORLD'S MOST COMPETITIVE RESTAURANT SERVICE COMPANY Year Ended February 29, 2016 CONTENTS 02 Financial and Non-financial Highlights 04 Message from the President and CEO 05 Business Strategy 08 Our Brands 10 Corporate Governance 12 Management’s Discussion and Analysis 14 Consolidated Financial Statements 17 Corporate Data Stock Code : 3387 What is create restaurants group? BASIC PHILOSOPHY CREATIVITY Since its foundation in 1999, create restaurants group has planned, developed, and operated restaurants in a wide variety of formats ranging from casual food courts and izakaya to restaurants offering a more formal dining experience. All these restaurants are attuned to SPEED CHALLENGE the characteristics of their locations and customer demographics, and the entire business is directly managed in accordance with an original strategy based on our fundamental philosophy of speed, creativity, and the pursuit of new challenges. Central to the mission of the create New York restaurants group is a concerted effort to earn the enduring trust of JAPAN our customers and develop new restaurant locations by drawing on SHANGHAI a wealth of experience and knowledge accumulated over the years. TAIWAN We will continue to expand our presence in Japan and abroad in the HONG KONG coming years. 197brands SLOGAN * SINGAPORE Becoming the world’s most competitive restaurant service company restaurants * Number of restaurants includes795 licensed businesses, franchised stores and overseas joint ventures. create restaurants inc. SFP Dining Co., Ltd. KR -

Prog-Web.Pdf
Programme Programme MARS - MAI ‘16 EXPOSITION MARS - MAI 2016 RÉTROSPECTIVE GUS VAN SANT RÉTROSPECTIVES HOU HSIAO-HSIEN Jean gabin Raoul ruiz Pierre richard MichÈle rosier Carte blanche à jean-michel alberola cinematheque.fr La Cinematheque-Parution MDE-012016 19/01/16 15:12 Page1 www.maisondesetatsunis.com POUR Vos voyages à PORTLAND La Maison des Etats-Unis ! Love Portland - 1670 €* Séjour de 7 j. / 5 n. Vols internationaux, hôtel*** Partez assisté d’une équipe de spécialistes qui fera de votre voyage un moment inoubliable. Retrouvez l’ensemble de nos offres www.maisondesetatsunis.com 3, rue Cassette Paris 6ème Tél. 01 53 63 13 43 [email protected] Du lundi au samedi de 10h à 19h. Prix à partir de, soumis à conditions Copyright Torsten Kjellstrand www.travelportland.com Gerry ÉDITORIAL Après Martin Scorsese, c’est à Gus Van Sant que la Cinémathèque française consacre une grande exposition et une rétrospective intégrale. Nourri de modernité euro- péenne, figure du cinéma américain indépendant dans les années 90, à l’instar d’un Jim Jarmush pour la décennie précédente, le cinéaste de Portland, Oregon est un authentique plasticien. Nous sommes heureux de montrer pour la première fois en France ses travaux de photographe et de peintre, et d’établir ainsi des correspon- dances avec ses films. Grand inventeur de dispositifs cinématographiques Elephant( , pour citer le plus connu de ses théorèmes), Gus Van Sant est aussi avide d’expériences commerciales, menées au cœur même de l’industrie (Prête-à-tout, Psychose, Will Hunting ou À la recherche de Forrester), et pour lesquelles il peut déployer toute sa plasticité d’auteur très américain, à la fois modeste et déterminé. -

Abstracts Pp. 152-450
Kingship Ideology in Sino-Tibetan Diplomacy during the VII-IX centuries Emanuela Garatti In this paper I would like to approach the question of the btsan-po’s figure and his role in the international exchanges like embassies, peace agreements and matrimonial alliances concluded between the Tibetan and the Tang during the Tibetan Empire. In order to do that, I examine some passages of Tibetan and Chinese sources. Tibetan ancient documents, like PT 1287, the PT 1288, the IOL Tib j 750 and the text of the Sino-Tibetan treaty of 821/822. For the Chinese sources I used the encyclopaedia Cefu yuangui which has never been extensively used in the study of the Tibetan ancient history. Concerning the embassies one can see that they are dispatched with important gifts when the btsan-po want to present a request. Those are registered as tribute (ch. chaogong) by the Chinese authors but one can assume, analysing the dates of embassies that the Tibetan emissaries are sent to the court with presents only when they had to present a specific request from the Tibetan emperor. Moreover, the btsan-po is willing to accept the diplomatic codes but refuses all attempt of submission from the Chinese authorities like the “fish-bag” (ch. yudai) proposed to the Tibetan ambassadors as a normal gift. For the treaties, the texts of these agreements show the evolution of the position of the btsan-po towards the Chinese court and the international diplomacy: the firsts pacts see the dominant position of Tang court over the btsan-po’s delegation. -

Kobe University Repository : Thesis
Kobe University Repository : Thesis Portugal's 'Estado Novo' Diplomatic Relations with Japan During the 学位論文題目 Second World War(第二次世界大戦間のポルトガルの「エスタド・ Title ノヴォ」と対日外交) 氏名 SOEIRO SIMOES DEBORA CATARINA Author 専攻分野 博士(政治学) Degree 学位授与の日付 2019-03-25 Date of Degree 公開日 2020-03-01 Date of Publication 資源タイプ Thesis or Dissertation / 学位論文 Resource Type 報告番号 甲第7394号 Report Number 権利 Rights JaLCDOI URL http://www.lib.kobe-u.ac.jp/handle_kernel/D1007394 ※当コンテンツは神戸大学の学術成果です。無断複製・不正使用等を禁じます。著作権法で認められている範囲内で、適切にご利用ください。 PDF issue: 2021-10-08 博士学位論文 論文題目 Portugal’s ‘Estado Novo’ Diplomatic Relations with Japan During the Second World War (第二次世界大戦間のポルトガルの「エスタド・ノヴォ」と対日外交) 神戸大学大学院法学研究科 専攻:政治学専攻 指導教授:簑原俊洋 学籍番号:141J035J 氏名:Débora Catarina Soeiro Simões 提出年月日: 2019 年 1 月 10 日 Abstract Portugal’s strategic dogma to survive the war with its colonial empire intact was easy to explain, but strenuous to implement: to publicly maintain a strict neutrality, but when inevitable to collaborate to protect its interests, thus the term collaborating neutrality. In reality this demanded a cautious and skillful use of hedging. Juggling the interests of the several actors was complex and sometimes out of the control of Portuguese authorities. Portuguese and Japanese interests collide in the Asia theater in Macau and East Timor. Both territories of strategic importance to Japan: the former as a espionage center and the latter as a stepping stone to Australia. In 1940, having Macau surrounded, Japan clearly states its intention to pressure Macau to obtain oil concessions in Timor. In turn, this precipitated a policy of appeasement on two fronts: in Macau, Portugal must cede part of its sovereignty in Macau, allowing Japanese a broad spectrum of freedoms; and in Timor, a technical agreement establishing an air service between Palao and Dilli is signed. -

Jam-Yang-Shay-Pa's Decisive Analysis of Dharmakīrti's
WHY DID DHARMAKĪRTI WRITE THE COMMENTARY? Jam-yang-shay-pa’s Decisive Analysis of Dharmakīrti’s “Commentary on Valid Cognition” Introduction 1 Hiroshi Nemoto In collaboration with Lo-sang-gyal-tshan Edited by Jeffrey Hopkins UMA INSTITUTE FOR TIBETAN STUDIES Why did Dharmakīrti Write the Commentary? Website for UMA Institute for Tibetan Studies (Union of the Modern and the Ancient: gsar rnying zung `jug khang): uma- tibet.org. UMA stands for “Union of the Modern and the Ancient” and means “Middle Way” in Tibetan. UMA is a non-profit 501(c)3 organization. Why did Dharmakīrti Write the Commentary? Jam-yang-shay-pa’s Decisive Analysis of Dharmakīrti’s “Commentary on Valid Cognition”: Introduction 1 Hiroshi Nemoto Edited by Jeffrey Hopkins UMA Institute for Tibetan Studies uma-tibet.org Expanding Wisdom and Compassion UMA Great Books Translation Project Supported by generous grants from the Yeshe Khorlo Foundation the Pierre and Pamela Omidyar Fund the Silicon Valley Community Foundation and a bequest from Daniel E. Perdue Translating texts from the heritage of Tibetan and Inner Asian Buddhist systems, the project focuses on the study and contemplation of Great Indian Books and Tibetan commentaries from the Go-mang College syllabus as well as a related theme on the fundamental innate mind of clear light in Tantric traditions. A feature of the Project is the usage of consistent vocabulary and format throughout the translations. Publications are available online without cost under a Creative Commons License with the understanding that downloaded mate- rial must be distributed for free: http://uma-tibet.org. UMA stands for Union of the Modern and the Ancient (gsar rnying zung ’jug khang). -
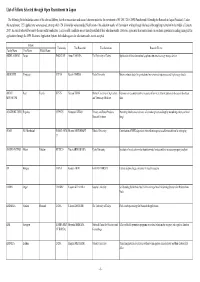
Under First Recruitment of FY 2017-2018 Program
List of Fellows Selected through Open Recruitment in Japan The following list includes the names of the selected fellows, their host researchers and research themes under the 1st recruitment of FY 2017-2018 JSPS Postdoctoral Fellowship for Research in Japan (Standard). Under this recruitment, 1221 applications were received, among which 124 fellowships were awarded. Notification of the selection results will be made in writing through the head of the applying institution in the middle of January, 2017. An award letter will be sent to the successful candidates. Unsuccessful candidates are not directly notified of their selection results. However, applicants (host researchers) can see their approximate ranking among all the applications through the JSPS Electronic Application System. Individual requests for selection results are not accepted. Fellow Nationality Host Researcher Host Institution Research Theme Family Name First Name Middle Name ABDUL SAMAD Yarjan PAKISTAN Atsuo YAMADA The University of Tokyo Application of three-dimensional graphene structure to energy storage devices ABOLIMITI Yimingaji CHINA Keiichi MAEDA Kyoto University Binary evoluton study for gravitational wave sources, supernovae and high energy objects ADJOU Paul Franck BENIN Xuenan XUAN Obihiro University of Agriculture Genomic survey and preventive measures of bovine tick-borne protozoan diseases in Southeast MOUMOUNI and Veterinary Medicine Asia AGATHOKLEOUS Evgenios CYPRUS Mitsutoshi KITAO Forestry and Forest Products Promoting abiotic stress tolerance of container-grown -

Annual Report Volume 21 Fiscal 2018
ISSN 1346-1192 Annual Report of the National Astronomical Observatory of Japan Volume 21 Fiscal 2018 Cover Caption This image shows the galaxy cluster MACS J1149.5+2223 taken with the NASA/ESA Hubble Space Telescope and the inset image is the galaxy MACS1149-JD1 located 13.28 billion light- years away observed with ALMA. Here, the oxygen distribution detected with ALMA is depicted in green. Credit: ALMA (ESO/NAOJ/NRAO), NASA/ESA Hubble Space Telescope, W. Zheng (JHU), M. Postman (STScI <http://www.stsci.edu/>), the CLASH Team, Hashimoto et al. Postscript Publisher National Institutes of Natural Sciences National Astronomical Observatory of Japan 2-21-1 Osawa, Mitaka-shi, Tokyo 181-8588, Japan TEL: +81-422-34-3600 FAX: +81-422-34-3960 https://www.nao.ac.jp/ Printer Kyodo Telecom System Information Co., Ltd. 4-34-17 Nakahara, Mitaka-shi, Tokyo 181-0005, Japan TEX: +81-422-46-2525 FAX: +81-422-46-2528 Annual Report of the National Astronomical Observatory of Japan Volume 21, Fiscal 2018 Preface Saku TSUNETA Director General I Scientific Highlights April 2018 – March 2019 001 II Status Reports of Research Activities 01. Subaru Telescope 048 02. Nobeyama Radio Observatory 053 03. Mizusawa VLBI Observatory 056 04. Solar Science Observatory (SSO) 061 05. NAOJ Chile Observatory (NAOJ ALMA Project / NAOJ Chile) 064 06. Center for Computational Astrophysics (CfCA) 067 07. Gravitational Wave Project Office 070 08. TMT-J Project Office 072 09. JASMINE Project Office 075 10. RISE (Research of Interior Structure and Evolution of Solar System Bodies) Project Office 077 11. Solar-C Project Office 078 12. -

From the OECD DAC Peer Review of Japan
Peer Review JAPAN Development Assistance Committee ORGANISATION FOR ECONOMIC CO-OPERATION AND DEVELOPMENT Peer Review of Japan ORGANISATION FOR ECONOMIC CO-OPERATION AND DEVELOPMENT Pursuant to Article 1 of the Convention signed in Paris on 14th December 1960, and which came into force on 30th September 1961, the Organisation for Economic Co-operation and Development (OECD) shall promote policies designed: - To achieve the highest sustainable economic growth and employment and a rising standard of living in member countries, while maintaining financial stability, and thus to contribute to the development of the world economy. - To contribute to sound economic expansion in member as well as non-member countries in the process of economic development. - To contribute to the expansion of world trade on a multilateral, non-discriminatory basis in accordance with international obligations. The original member countries of the OECD are Austria, Belgium, Canada, Denmark, France, Germany, Greece, Iceland, Ireland, Italy, Luxembourg, the Netherlands, Norway, Portugal, Spain, Sweden, Switzerland, Turkey, the United Kingdom and the United States. The following countries became members subsequently through accession at the dates indicated hereafter: Japan (28th April 1964), Finland (28th January 1969), Australia (7th June 1971), New Zealand (29th May 1973), Mexico (18th May 1994), the Czech Republic (21st December 1995), Hungary (7th May 1996), Poland (22nd November 1996), Korea (12th December 1996) and the Slovak Republic (14th December 2000). The Commission of the European Communities takes part in the work of the OECD (Article 13 of the OECD Convention). In order to achieve its aims the OECD has set up a number of specialised committees. -

(Jang-Kya Röl-Pay-Dor-Je's) “Song of the View, Identifying Mama”
Kön-chog-jig-me-wang-po’s Commentary on (Jang-kya Röl-pay-dor-je’s) “Song of the View, Identifying Mama”: Lamp for the Words Translated and Annotated by Jeffrey Hopkins UMA INSTITUTE FOR TIBETAN STUDIES “Identifying Mama”: Lamp for the Words Website for UMA Institute for Tibetan Studies (Union of the Modern and the Ancient: gsar rnying zung `jug khang): uma- tibet.org. UMA stands for "Union of the Modern and the Ancient" and means "Middle Way" in Tibetan. UMA is a non-profit 501(c)3 charity. For His Holiness Dalai Lama He has inspired the re-building of Tibetan cultural institutions outside of Tibet. He has asked the religious and the political leaders of the world to look beyond narrow interests to the greater good. He has advocated atten- tion to the basic need of society regardless of religion or politics—kind- ness. Is this what is feared? His power comes from a life of ethics, the force of truth. In Tibetan, he speaks with a range, depth, inspiration, humor, and sincerity that inspire insight and motivate dedication to others’ welfare. I have often wished that all the world could hear this marvel in his own tongue. His longer name rJe btsun 'jam dpal ngag dbang blo bzang ye shes bstan 'dzin rgya mtsho srid gsum dbang bsgyur mtshungs pa med pa'i sde dpal bzang po, which in English syllable-by-syllable is “Leader, holiness, gentleness, renown, speech, dominion, mind, goodness, primordial, wis- dom, teaching, hold, vastness, ocean, being, triad, controlling, unparal- leled, glory, integrity.” Leader of the world recognized for true holiness, -

GIP-Gip2019ces-1UP 1..179
CANCERS OF THE ESOPHAGUS AND STOMACH 2 Oral Abstract Session, Thu, 2:15 PM-3:45 PM and Poster Session (Board #D12), Thu, 11:30 AM-1:00 PM and 5:30 PM-6:30 PM Pembrolizumab versus chemotherapy as second-line therapy for advanced esophageal cancer: Phase III KEYNOTE-181 study. Takashi Kojima, Kei Muro, Eric Francois, Chih-Hung Hsu, Toshikazu Moriwaki, Sung-Bae Kim, Se-Hoon Lee, Jaafar Bennouna, Ken Kato, Shen Lin, Shi-Qui Qin, Paula Ferreira, Toshihiko Doi, Antoine Adenis, Peter C. Enzinger, Manish A. Shah, Ruixue Wang, Pooja Bhagia, S. Peter Kang, Jean-Philippe Metges; Department of Gastroenterology and Gastrointestinal Oncology, National Cancer Center Hospital East, Kashiwa, Japan; Department of Clinical Oncology, Aichi Cancer Center Hospital, Nagoya, Japan; Lacassagne Anticancer Center, Nice, France; National Taiwan University Hospital, Taipei, Taiwan; University of Tsukuba, Tsukuba, Japan; Department of Oncology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea; Division of Hematology and Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea; University Hospital of Nantes, Nantes, France; Division of Gastrointestinal Medical Oncology, National Cancer Center Hospital, Tokyo, Japan; Beijing Cancer Hospital, Beijing, China; PLA Cancer Centre of Nanjing Bayi Hospital, Nanjing, China; Instituto Portuguesˆ de Oncologia Porto, Porto, Portugal; National Cancer Center Hospital East, Chiba, Japan; Centre Oscar Lambret, Lille, France; Dana-Farber Cancer Institute, Boston, MA; Weill Cornell Medicine/ New York Presbyterian Hospital, New York, NY; MSD China, Beijing, China; Merck, Kenilworth, NJ; Merck & Co., Inc., Kenilworth, NJ; Oncology & Haematology Institute, Brest University Hospital, Brest, France Background: Patients with advanced esophageal cancer after first-line chemotherapy (chemo) have a poor prognosis and limited treatment options. -

Kobe University Repository : Thesis
Kobe University Repository : Thesis Portugal's 'Estado Novo' Diplomatic Relations with Japan During the 学位論文題目 Second World War(第二次世界大戦間のポルトガルの「エスタド・ Title ノヴォ」と対日外交) 氏名 SOEIRO SIMOES DEBORA CATARINA Author 専攻分野 博士(政治学) Degree 学位授与の日付 2019-03-25 Date of Degree 公開日 2020-03-01 Date of Publication 資源タイプ Thesis or Dissertation / 学位論文 Resource Type 報告番号 甲第7394号 Report Number 権利 Rights JaLCDOI URL http://www.lib.kobe-u.ac.jp/handle_kernel/D1007394 ※当コンテンツは神戸大学の学術成果です。無断複製・不正使用等を禁じます。著作権法で認められている範囲内で、適切にご利用ください。 PDF issue: 2020-04-09 博士学位論文 論文題目 Portugal’s ‘Estado Novo’ Diplomatic Relations with Japan During the Second World War (第二次世界大戦間のポルトガルの「エスタド・ノヴォ」と対日外交) 神戸大学大学院法学研究科 専攻:政治学専攻 指導教授:簑原俊洋 学籍番号:141J035J 氏名:Débora Catarina Soeiro Simões 提出年月日: 2019 年 1 月 10 日 Abstract Portugal’s strategic dogma to survive the war with its colonial empire intact was easy to explain, but strenuous to implement: to publicly maintain a strict neutrality, but when inevitable to collaborate to protect its interests, thus the term collaborating neutrality. In reality this demanded a cautious and skillful use of hedging. Juggling the interests of the several actors was complex and sometimes out of the control of Portuguese authorities. Portuguese and Japanese interests collide in the Asia theater in Macau and East Timor. Both territories of strategic importance to Japan: the former as a espionage center and the latter as a stepping stone to Australia. In 1940, having Macau surrounded, Japan clearly states its intention to pressure Macau to obtain oil concessions in Timor. In turn, this precipitated a policy of appeasement on two fronts: in Macau, Portugal must cede part of its sovereignty in Macau, allowing Japanese a broad spectrum of freedoms; and in Timor, a technical agreement establishing an air service between Palao and Dilli is signed.