Network Dynamics Determine the Autocrine and Paracrine Signaling Functions of TNF
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Met Receptor Tyrosine Kinase: Enhanced Signaling Through Adapter Proteins
Oncogene (2000) 19, 5582 ± 5589 ã 2000 Macmillan Publishers Ltd All rights reserved 0950 ± 9232/00 $15.00 www.nature.com/onc Met receptor tyrosine kinase: enhanced signaling through adapter proteins Kyle A Furge1, Yu-Wen Zhang1 and George F Vande Woude*,1 1Van Andel Research Institute, 333 Bostwick, N.E., Grand Rapids, Michigan, MI 49503, USA The Met receptor tyrosine kinase is the prototypic matrix (`invasion') (Jeers et al., 1996c; Matsumoto et member of a small subfamily of growth factor receptors al., 1994; Rong et al., 1994; Weidner et al., 1990). In that when activated induce mitogenic, motogenic, and addition, HGF/SF-Met signaling can induce several morphogenic cellular responses. The ligand for Met is dierent epithelial and mesenchymal cell types to hepatocyte growth factor/scatter factor (HGF/SF) and undergo an involved dierentiation program termed while normal HGF/SF-Met signaling is required for branching morphogenesis when the cells are grown in a embryonic development, abnormal Met signaling has three dimensional matrix (Brinkmann et al., 1995; been strongly implicated in tumorigenesis, particularly in Jeers et al., 1996c; Montesano et al., 1991a; Niemann the development of invasive and metastatic phenotypes. et al., 1998). During branching morphogenesis, groups Following ligand binding and autophosphorylation, Met of cells proliferate, migrate, and dierentiate to form a transmits intercellular signals using a unique multi- connected series of tubules arranged like branches from substrate docking site present within the C-terminal a tree. However, even in the absense of a three end of the receptor. The multisubstrate docking site dimensional matrix, signaling through the Met receptor mediates the binding of several adapter proteins such as can induce morphogenesis and lumen formation in Grb2, SHC, Crk/CRKL, and the large adapter protein certain cell types (Jeers et al., 1996a; Tsarfaty et al., Gab1. -

Paracrine Wnt1 Drives Interstitial Fibrosis Without Inflammation by Tubulointerstitial Cross-Talk
BASIC RESEARCH www.jasn.org Paracrine Wnt1 Drives Interstitial Fibrosis without Inflammation by Tubulointerstitial Cross-Talk † Omar H. Maarouf,* Anusha Aravamudhan,* Deepika Rangarajan,* Tetsuro Kusaba,* ‡ Victor Zhang,* Jeremy Welborn,* Daniel Gauvin,* Xiuyun Hou,* Rafael Kramann,* and † Benjamin D. Humphreys* § *Renal Division, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts; †Harvard Medical School, Boston, Massachusetts; ‡Division of Nephrology and Clinical Immunology and Medical Faculty, Rheinisch- Westfälische Technische Hochschule Aachen University, Aachen, Germany; and §Harvard Stem Cell Institute, Cambridge, Massachusetts ABSTRACT AKI with incomplete epithelial repair is a major contributor to CKD characterized by tubulointerstitial fibrosis. Injury–induced epithelial secretion of profibrotic factors is hypothesized to underlie this link, but the identity of these factors and whether epithelial injury is required remain undefined. We previously showed that activation of the canonical Wnt signaling pathway in interstitial pericytes cell autonomously drives myofibroblast acti- vation in vivo. Here, we show that inhibition of canonical Wnt signaling also substantially prevented TGFb– dependent myofibroblast activation in vitro. To investigate whether Wnt ligand derived from proximal tubule is sufficient for renal fibrogenesis, we generated a novel mouse strain with inducible proximal tubule Wnt1 secretion. Adult mice were treated with vehicle or tamoxifen and euthanized at 12 or 24 weeks postinjection. Compared -

Paracrine Signaling Mediated at Cellcell Contacts
Insights & Perspectives Think again Paracrine signaling mediated at cellÀcell contacts Sougata Roy*,† and Thomas B. Kornberg Recent findings in several organ systems show that cytoneme-mediated systems. However, recent work that we signaling transports signaling proteins along cellular extensions and targets discuss here describes paracrine signal- cell-to-cell exchanges to synaptic contacts. This mechanism of paracrine ing that is instead contact-mediated and signaling may be a general one that is used by many (or all) cell types in many (or dependent on transient synapses that all) organs. We briefly review these findings in this perspective. We also non-neuronal cells make. These synap- describe the properties of several signaling systems that have previously been ses form at sites where specialized signaling filopodia called cytonemes interpreted to support a passive diffusion mechanism of signaling protein extend to contact target cells. dispersion, but can now be understood in the context of the cytoneme mechanism. Keywords: The classical model of .cytonemes; filopodia; morphogen; paracrine signaling; synapse; TGF-b paracrine signaling assumes that signals disperse by passive Introduction so that signals are within only 15À20 nm diffusion of their target receptors when they are Animal cells communicate over long released. Paracrine signaling, the third There are many paracrine signaling distances in various ways. Endocrine general mechanism, may be considered proteins that have been characterized. cells signal systemically by releasing to be a variant of endocrine signaling, They include the Fibroblast Growth hormones that disseminate in the vas- functioning at relatively short range Factors (FGFs) and other proteins that culature. Neurons also release signals, when secreted signals move limited activate Receptor Tyrosine Kinases, but they exchange information at syn- distances by passive diffusion in extra- TGF-b family members, Wnt proteins, apses that form where their axons and cellular fluid. -

Cell Communication and Signaling Mechanisms Terms to Learn
LQB383 Testbank Week 8 – Cell Communication and Signaling Mechanisms Terms to learn – match the terms to the definitions -------------------------------------------------------------------------------------------------------------------------- Adaptor Intracellular signaling protein Morphogen Endocrine cell Gap junction Neurotransmitter Hormone Receptor Extracellular signal molecule Signaling cascade Steroid hormone Synaptic signaling Paracrine signaling Second messenger Autocrine signaling Nuclear receptor family Interaction domain Contact-dependent signaling Definitions 1. General term for a protein that binds a specific extracellular molecule (ligand) and initiates a response in a cell. 2. Communicating cell-cell junction that allows ions and small molecules to pass from the cytoplasm of one cell to the cytoplasm of another. 3. Hydrophobic signaling molecule with characteristic four-ringed structure derived from cholesterol. 4. Short-range cell-cell communication via secreted local mediators that act on adjacent cells. 5. Specialised cell that secretes a hormone into the blood 6. The cell responds to its own secreted molecules. 7. Small signaling molecule secreted by the presynaptic nerve cell at a chemical synapse to relay the signal to the postsynaptic cell. 8. Small molecule that is formed in the cytosol, or released into it, in response to an extracellular signal, and that helps to relay the signal in the interior of the cell. 9. Molecule from the outside of the cell that communicates the behaviour or actions of other cells in the environment and elicits an appropriate response. 10. General term for a signal relay chain containing multiple amplification steps. 1 Receptor 2 Gap junction 3 Steroid hormone 4 Paracrine signaling 5 Endocrine cell 6 Autocrine signaling 7 Neurotransmitter 8 Second messenger 9 Extracellular signal molecule 10 Signaling cascade Multiple Choice Questions 1. -

Paracrine Signaling by Progesterone ⇑ Renuga Devi Rajaram, Cathrin Brisken
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Infoscience - École polytechnique fédérale de Lausanne Molecular and Cellular Endocrinology xxx (2011) xxx–xxx Contents lists available at SciVerse ScienceDirect Molecular and Cellular Endocrinology journal homepage: www.elsevier.com/locate/mce Review Paracrine signaling by progesterone ⇑ Renuga Devi Rajaram, Cathrin Brisken Ecole Polytechnique Fédérale de Lausanne (EPFL), ISREC – Swiss Institute for Experimental Cancer Research, NCCR Molecular Oncology, SV2832 Station 19, CH-1015 Lausanne, Switzerland article info abstract Article history: Steroid hormones coordinate and control the development and function of many organs and are impli- Available online xxxx cated in many pathological processes. Progesterone signaling, in particular, is essential for several impor- tant female reproductive functions. Physiological effects of progesterone are mediated by its cognate Keywords: receptor, expressed in a subset of cells in target tissues. Experimental evidence has accumulated that pro- Progesterone receptor gesterone acts through both cell intrinsic as well as paracrine signaling mechanisms. By relegating the Paracrine signaling hormonal stimulus to paracrine signaling cascades the systemic signal gets amplified locally and signal- Uterus ing reaches different cell types that are devoid of hormone receptors. Interestingly, distinct biological Ovaries responses to progesterone in different target tissues rely on several tissue-specific and some common Mammary gland Carcinogenesis paracrine factors that coordinate biological responses in different cell types. Evidence is forthcoming that the intercellular signaling pathways that control development and physiological functions are important in tumorigenesis. Crown Copyright Ó 2011 Published by Elsevier Ireland Ltd. All rights reserved. Contents 1. Introduction . ....................................................................................................... 00 2. -

Paracrine-Induced Response State Antiviral-Activated Dendritic Cells: A
Antiviral-Activated Dendritic Cells: A Paracrine-Induced Response State Antonio V. Bordería, Boris M. Hartmann, Ana Fernandez-Sesma, Thomas M. Moran and Stuart C. Sealfon This information is current as of September 24, 2021. J Immunol 2008; 181:6872-6881; ; doi: 10.4049/jimmunol.181.10.6872 http://www.jimmunol.org/content/181/10/6872 Downloaded from References This article cites 53 articles, 22 of which you can access for free at: http://www.jimmunol.org/content/181/10/6872.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 24, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2008 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Antiviral-Activated Dendritic Cells: A Paracrine-Induced Response State1 Antonio V. Bordería,2* Boris M. Hartmann,2† Ana Fernandez-Sesma,* Thomas M. Moran,* and Stuart C. Sealfon3†‡ Infection of immature dendritic cells (DCs) by virus stimulates their maturation into APC. -

Hedgehog Signaling: Networking to Nurture a Promalignant Tumor Microenvironment
Published OnlineFirst July 20, 2011; DOI: 10.1158/1541-7786.MCR-11-0175 Molecular Cancer Subject Review Research Hedgehog Signaling: Networking to Nurture a Promalignant Tumor Microenvironment Lillianne G. Harris, Rajeev S. Samant, and Lalita A. Shevde Abstract In addition to its role in embryonic development, the Hedgehog pathway has been shown to be an active participant in cancer development, progression, and metastasis. Although this pathway is activated by autocrine signaling by Hedgehog ligands, it can also initiate paracrine signaling with cells in the microenvironment. This creates a network of Hedgehog signaling that determines the malignant behavior of the tumor cells. As a result of paracrine signal transmission, the effects of Hedgehog signaling most profoundly influence the stromal cells that constitute the tumor microenvironment. The stromal cells in turn produce factors that nurture the tumor. Thus, such a resonating cross-talk can amplify Hedgehog signaling, resulting in molecular chatter that overall promotes tumor progression. Inhibitors of Hedgehog signaling have been the subject of intense research. Several of these inhibitors are currently being evaluated in clinical trials. Here, we review the role of the Hedgehog pathway in the signature characteristics of cancer cells that determine tumor development, progression, and metastasis. This review condenses the latest findings on the signaling pathways that are activated and/or regulated by molecules generated from Hedgehog signaling in cancer and cites promising clinical interventions. Finally, we discuss future directions for identifying the appropriate patients for therapy, developing reliable markers of efficacy of treatment, and combating resistance to Hedgehog pathway inhibitors. Mol Cancer Res; 9(9); 1165–74. -

Inflammatory Stimuli Differentially Modulate the Transcription of Paracrine Signaling Molecules of Equine Bone Marrow Multipoten
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Osteoarthritis and Cartilage 21 (2013) 1116e1124 Inflammatory stimuli differentially modulate the transcription of paracrine signaling molecules of equine bone marrow multipotent mesenchymal stromal cells R. Vézina Audette, A. Lavoie-Lamoureux, J.-P. Lavoie, S. Laverty* Comparative Orthopedic Research Laboratory, Département de sciences cliniques, Faculté de Médecine vétérinaire, Université de Montréal, St Hyacinthe, QC, Canada article info summary Article history: Objective: Osteoarthritis (OA) is a degenerative disease of joint tissues that causes articular cartilage Received 30 November 2012 erosion, osteophytosis and loss of function due to pain. Inflammation and inflammatory cytokines in Accepted 3 May 2013 synovial fluid (SF) contribute to OA progression. Intra-articular (IA) injections of multipotent mesen- chymal stromal cells (MSCs) are employed to treat OA in both humans and animals. MSCs secrete Keywords: paracrine pro-inflammatory and anabolic signaling molecules that promote tissue repair. The objective of Equine this study was to investigate the effects of OASF on the gene expression of paracrine signaling molecules Mesenchymal stem cells by MSCs. Multipotent mesenchymal stromal cells b Paracrine communication Methods: The effects of Lipopolysaccharide (LPS) and interleukin (IL)-1 as well as both normal (N) and fl Synovial fluid osteoarthritis (OA) SF stimulations on the expression of paracrine pro-in ammatory (tumor necrosis Reverse transcriptase polymerase chain factor (TNF)-a, IL-1b, IL-8), modulatory (IL-6) and anabolic (vascular endothelial growth factor (VEGF), reaction transforming growth factor (TGF)-b1 and insulin-like growth factor (IGF)-1) signaling molecules by Osteoarthritis equine bone marrow multipotent mesenchymal stromal cells (eBM-MSCs) was investigated employing Cartilage reverse transcriptase-polymerase chain reaction (RT-PCR). -

The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor
International Journal of Molecular Sciences Review The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor Annalisa Astolfi 1 , Maria Abbondanza Pantaleo 2,*, Valentina Indio 3 , Milena Urbini 4 and Margherita Nannini 5 1 Department of Morphology, Surgery and Experimental Medicine, University of Ferrara, 44121 Ferrara, Italy; annalisa.astolfi@unife.it 2 Department of Experimental, Diagnostic and Specialty Medicine, University of Bologna, 40138 Bologna, Italy 3 “Giorgio Prodi” Cancer Research Center, University of Bologna, 40138 Bologna, Italy; [email protected] 4 Biosciences Laboratory, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS, 47014 Meldola, Italy; [email protected] 5 Medical Oncology Unit, S.Orsola-Malpighi University Hospital, 40138 Bologna, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-051-214-4043; Fax: +39-051-349-655 Received: 10 April 2020; Accepted: 4 May 2020; Published: 7 May 2020 Abstract: Gastrointestinal stromal tumors (GIST) are rare neoplasms of mesenchymal origin arising in the gastrointestinal tract. The vast majority are characterized by mutually exclusive activating mutations in KIT or Platelet-derived growth factor alpha (PDGFRA) receptors, or less frequently by succinate dehydrogenase complex (SDH) or NF1 inactivation, with very rare cases harboring mutant BRAF or RAS alleles. Approximately 5% of GISTs lack any of such mutations and are called quadruple wild-type (WT) GISTs. Recently, deregulated Fibroblast Growth Factor (FGF)/FGF-receptor (FGFR) signaling emerged as a relevant pathway driving oncogenic activity in different molecular subgroups of GISTs. This review summarizes all the current evidences supporting the key role of the FGF/FGFR pathway activation in GISTs, whereby either activating mutations, oncogenic gene fusions, or autocrine/paracrine signaling have been detected in quadruple WT, SDH-deficient, or KIT-mutant GISTs. -

Chapter 11 Cell Communication
Chapter 11 Cell Communication PowerPoint® Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from Joan Sharp Copyright © 2008 Pearson Education, Inc., publishing as Pearson Benjamin Cummings Overview: The Cellular Internet • Cell-to-cell communication is essential for multicellular organisms • Biologists have discovered some universal mechanisms of cellular regulation • The combined effects of multiple signals determine cell response • For example, the dilation of blood vessels is controlled by multiple molecules Copyright © 2008 Pearson Education, Inc., publishing as Pearson Benjamin Cummings Fig. 11-1 Concept 11.1: External signals are converted to responses within the cell • Microbes are a window on the role of cell signaling in the evolution of life Copyright © 2008 Pearson Education, Inc., publishing as Pearson Benjamin Cummings Evolution of Cell Signaling • A signal transduction pathway is a series of steps by which a signal on a cell’s surface is converted into a specific cellular response • Signal transduction pathways convert signals on a cell’s surface into cellular responses Copyright © 2008 Pearson Education, Inc., publishing as Pearson Benjamin Cummings Fig. 11-2 factor Receptor 1 Exchange a of mating factors a factor Yeast cell, Yeast cell, mating type a mating type 2 Mating a 3 New a/ a/ cell • Pathway similarities suggest that ancestral signaling molecules evolved in prokaryotes and were modified later in eukaryotes • The -

Paracrine Signaling from a Three-Dimensional Model of Bladder Carcinoma and from Normal Bladder Switch the Phenotype of Stromal Fibroblasts
cancers Article Paracrine Signaling from a Three-Dimensional Model of Bladder Carcinoma and from Normal Bladder Switch the Phenotype of Stromal Fibroblasts Sandra Camargo 1, Ofer N. Gofrit 2, Assaf Assis 1 and Eduardo Mitrani 1,* 1 Department of Cell and Developmental Biology, The Hebrew University of Jerusalem, Givat Ram, Jerusalem 91904, Israel; [email protected] (S.C.); [email protected] (A.A.) 2 Department of Urology, Hadassah Medical Center, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem 91120, Israel; [email protected] * Correspondence: [email protected]; Tel.: +972-26585925; Fax: +972-26585660 Simple Summary: The aim of this work was to create a bladder carcinoma model to study the signals secreted by carcinoma cells in a in vivo-like context and how this molecular signaling can alter stromal fibroblasts. Our model is based on decellularized organ fragments (scaffolds) that aim to preserve the complexity of the native extracellular matrix of the bladder. Primary epithelial cells isolated from human bladder carcinoma were seeded on the scaffolds. We found that those cells showed a similar gene expression pattern when seeded on the scaffolds or on monolayer cultures. However, the secreted pattern of key growth factors was significantly different. Only the combination of factors secreted by carcinoma cells seeded on the scaffolds, but not the carcinoma cells seeded on plastic, was able to induce a pro-inflammatory or myofibroblast phenotype. This model allows one Citation: Camargo, S.; Gofrit, O.N.; to decipher the paracrine pathways of bladder carcinoma in a defined in vitro system. -
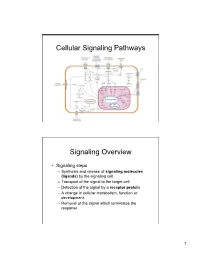
Cellular Signaling Pathways
Cellular Signaling Pathways Signaling Overview • Signaling steps – Synthesis and release of signaling molecules (ligands) by the signaling cell. – Transport of the signal to the target cell – Detection of the signal by a receptor protein – A change in cellular metabolism, function or development. – Removal of the signal which terminates the response 1 Signaling Overview • Endocrine - hormones act on target cells which are distant from their site of synthesis. (examples: testosterone, estrogen, thyroid hormone….) • Paracrine - Signaling molecules released by a cell only affect those cells in close proximity to it. (examples: neurotransmitters, growth factors) Signaling Overview • Autocrine signaling - cells respond to substances they themselves release. (examples: growth factors and T-lymphocytes) • Membrane bound proteins- proteins on one cell can directly signal an adjacent cell. (example: embryo development) 2 Signaling Overview • Different cell types may have different sets of receptors for the same ligand each of which induces a different response. • The same receptor may occur on different cell types and binding of the same ligand may invoke a different response from each cell type. • Different ligand-receptor complexes can induce the same cellular response in some cell types. Four Classes of Receptors • G-protein coupled receptors – Ligand binding activates a G-protein which in turn activates or inhibits an enzyme that generates a secondary messenger or modulates an ion channel. – Examples: receptors for epinephrine, serotonin and glucagon, light activated receptors, receptors for neurotransmitters. 3 Four Classes of Receptors • Ion channel receptors – Ligand binding changes the conformation of the receptor to allow the movement of ions across the membrane – Example: receptors for acetylcholine Four Classes of Receptors • Tyrosine kinase linked receptors – Protein kinases: An enzyme which phosphorylates specific serine, threonine or tyrosine residues in target proteins.