UC Irvine UC Irvine Electronic Theses and Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Your Family's Guide to Explore NYC for FREE with Your Cool Culture Pass
coolculture.org FAMILY2019-2020 GUIDE Your family’s guide to explore NYC for FREE with your Cool Culture Pass. Cool Culture | 2019-2020 Family Guide | coolculture.org WELCOME TO COOL CULTURE! Whether you are a returning family or brand new to Cool Culture, we welcome you to a new year of family fun, cultural exploration and creativity. As the Executive Director of Cool Culture, I am excited to have your family become a part of ours. Founded in 1999, Cool Culture is a non-profit organization with a mission to amplify the voices of families and strengthen the power of historically marginalized communities through engagement with art and culture, both within cultural institutions and beyond. To that end, we have partnered with your child’s school to give your family FREE admission to almost 90 New York City museums, historic societies, gardens and zoos. As your child’s first teacher and advocate, we hope you find this guide useful in adding to the joy, community, and culture that are part of your family traditions! Candice Anderson Executive Director Cool Culture 2020 Cool Culture | 2019-2020 Family Guide | coolculture.org HOW TO USE YOUR COOL CULTURE FAMILY PASS You + 4 = FREE Extras Are Extra Up to 5 people, including you, will be The Family Pass covers general admission. granted free admission with a Cool Culture You may need to pay extra fees for special Family Pass to approximately 90 museums, exhibits and activities. Please call the $ $ zoos and historic sites. museum if you’re unsure. $ More than 5 people total? Be prepared to It’s For Families pay additional admission fees. -

Sicilian Eatery
SICILIAN EATERY Concrete is a casual dining experience located on the border of Brooklyn neighborhoods Bedford Stuyvesant + Bushwick. 917-886-9660 The 3,000 sq ft space has a capacity of 75, featuring an open kitchen, dining area, full bar with seating, and performance stage. 906 Broadway, Concrete’s menu includes a variety of plates from both Italian and American cuisines, Brooklyn NY 11206 in addition to a selection of Sicilian street food favorites. 906broadway The bar offers a selection of top shelf liquors, Sicilian wines, and local beers. @gmail.com The list of specialty cocktails are curated exclusively for Concrete, with all cocktail syrups made fresh + in house by our bartending staff. @concretebrooklyn Open June 2018, the space features artwork from both local and international artists, www. including a custom mural from artist Mike Lee on the building’s Stockton St side. concrete-brooklyn The live event calendar is set to premiere in Fall 2018. .com Currently serving dinner from 5p - 11p, with brunch available on weekends. Located at 906 Broadway, Brooklyn NY 11206, accessible by the J/M/Z trains at Myrtle-Broadway + the B46 bus. THE NEAREST TRAINS ARE THE M/J/Z LINES AT MYRTLE - BROADWAY, OR FLUSHING AVE. WE ARE A 14 MINUTE WALK FROM THE HALSEY L TRAIN STATION IN BUSHWICK. THE B46, B47, B54, M1, M119 (AT MYRTLE AVE), AND B15 (AT LEWIS AVE) HAVE ROUTES TO MYRTLE / BROADWAY JUNCTION, AND ARE WITHIN A 3-10 MINUTE WALK FROM CONCRETE’S LOCATION. High resolution images can be downloaded here: https://www.dropbox.com/sh/1upry4klu8zc8ll/AADVl9DsBLjDfT5-E4H1iezNa?dl=0 Interior and Exterior Space Photography: Leonardo Mascaro http://www.leonardomascaro.com Food + Drink Photography: Paul Quitoriano http://www.paulcrispin.com DOP GRADE PRODUCTS Dnominazione di Origine Protetta certification ensures that products are locally grown and packaged. -

Fixing the L Train and Managing the Shutdown a Community Consensus Proposal
Fixing the L Train and Managing the Shutdown A Community Consensus Proposal November 2016 Contents Executive Summary / 3 Summary of Recommendations / 3 Introduction / 6 Impact on Commuters and Residents / 8 Implications/how to prepare for the shutdown / 10 Impact on Businesses / 11 How much do local businesses depend on the L train? / 11 How to prepare for the shutdown / 11 Providing the Best Travel Alternatives / 12 Prepare adjacent subway lines for higher ridership / 12 New rapid bus services with dedicated preferential treatments and auto-free zones / 13 Transform streets in Brooklyn to better connect people and cyclists to transit / 17 Improve ferry service and reduce fares to serve Williamsburg residents / 18 Making the Most of the Shutdown: Transforming the L Train / 19 Capital improvements at five stations / 20 Timing and funding / 20 Procurement and design / 21 An Inclusive Process / 22 Community Profiles /23 Manhattan / 24 Williamsburg/Greenpoint / 25 Bushwick/Ridgewood / 26 East New York/Brownsville/Canarsie / 27 2 Fixing the L Train and Managing the Shutdown: A Community Consensus Proposal | November 2016 Executive Summary The Metropolitan Transportation Authority has said it will shut ⊲ State Senator Martin M. Dilan down the L train tunnels under the East River for more than a ⊲ Council Member Stephen Levin year to repair the severe damage caused by Superstorm Sandy. ⊲ Council Member Antonio Reynoso That is grim news for the hundreds of thousands of New Yorkers ⊲ Manhattan Borough President Gale Brewer who rely on the L and who will have few easy alternatives to get ⊲ Brooklyn Borrough President Eric L. Adams to where they’re going every day. -

Too Cool—Families Catch the Cool!
2010 SPRING Cool Culture® provides 50,000 underserved families with free, unlimited sponsored by JAQUELINE KENNEDY access to ONASSIS 90 cultural institutionsRESEVOIR - so that parents can provide their children withCENTRAL PARK 80 Hanson Place, Suite 604, Brooklyn, NY 11217 www.coolculture.org educational experiences that will help them succeed in school and life. CENTRAL PARK HARLEM MEER Malky, Simcha, Stanley and Avi Mayerfeld. Fi e tzpa t trick t . Vaness e a Griffi v th and Ys Y abe l Fitzpat FIFTH AVENUE d rick. n a o FIFTH AVENUE i g r e S , a n i t n e g r A Isabella, Sophia and Ethel Zaldaña 108TH ST 107TH ST 106TH ST 103RD ST 105TH ST 102ND ST 104TH ST 101ST ST 100TH ST 99TH ST 98TH ST 97TH ST 96TH ST 95TH ST 94TH ST 93RD ST 92ND ST 91ST ST 90TH ST 89TH ST 88TH ST 87TH ST 86TH ST 85TH ST 84TH ST 83RD ST 82ND ST 81ST ST Felicia and Omaria Williams F e l ic ia a nd he t C C O o o m o a h ri W o To ol— illiams atc l! Families C The Cool Culture community couldn't choose just one. “I really liked came together to Catch the Cool on making stuff and meeting my friend and June 8th at the Museum Mile getting a poster by (artist) Michael Albert,” she said. The siblings – along with Festival! Thousands painted, drew, their sister Ysabel (one), mom Yvette and aunt danced and partied on Fifth Avenue from Vanessa Griffith– participated in art activities 105th Street to 82nd Street, dropping in that included crafting monkey ears at The museums along the way. -

Dnalc-Nyc-City-Tech-Directions.Pdf
Mail to: 1 Bungtown Road Cold Spring Harbor, NY 11724 Phone: (516) 367-5170 Fax: (516) 367-5182 Internet: dnalc.cshl.edu Email: [email protected] DIRECTIONS DNALC NYC at City Tech Enter at: 259 Adams Street, Brooklyn, NY 11201 (516) 719-1296 (Alternative: 516-367-5170) By subway: 2, 3, 4, 5: Get off at the Borough Hall station and walk towards Borough Hall to 259 Adams St. A, C, F, R: Get off at Jay Street-Metro Tech station and walk towards Borough Hall to 259 Adams St. MTA Subway map: http://www.mta.info/nyct/maps/submap.htm By bus: B26, B37, B38, B41, B51, B52, B54, B61, B67 or B75. MTA Bus Map: https://new.mta.info/maps/bus Driving Directions: Manhattan/Bronx: FDR Drive to the Brooklyn Bridge. When you exit the bridge the DNALC NYC at City Tech is just past Tillary Street on the left, drop off is 259 Adams Street entrance. Queens: Long Island Expressway west to the BQE. Follow the sign to downtown Brooklyn. Exit at Tillary and stay in the center lane past the Flatbush Avenue Extensions; 3 more blocks and turn left on Tillary. DNALC NYC at City Tech will be on the left, drop off is 259 Adams Street entrance. Brooklyn: Belt Parkway to the Brooklyn-Queens Expressway and get off at the Atlantic Avenue exit. Turn right on Atlantic; continue for five blocks to Smith Street. Make a left onto Smith; continue across Fulton where Smith becomes Jay. Make left on Johnson Street, and a right onto Adams St/Brooklyn Bridge Blvd. -

Improving Bus Service in New York a Thesis Presented to The
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Columbia University Academic Commons Improving Bus Service in New York A Thesis Presented to the Faculty of Architecture and Planning COLUMBIA UNIVERSITY In Partial Fulfillment Of the requirements for the Degree Master of Science in Urban Planning By Charles Romanow May 2018 Abstract New York City’s transportation system is in a state of disarray. City street are clogged with taxi’s and for-hire vehicles, subway platforms are packed with straphangers waiting for delayed trains and buses barely travel faster than pedestrians. The bureaucracy of City and State government in the region causes piecemeal improvements which do not keep up with the state of disrepair. Bus service is particularly poor, moving at rates incomparable with the rest of the country. New York has recently made successful efforts at improving bus speeds, but only so much can be done amidst a city of gridlock. Bus systems around the world faced similar challenges and successfully implemented improvements. A toolbox of near-immediate and long- term options are at New York’s disposal dealing directly with bus service as well indirect causes of poor bus service. The failing subway system has prompted public discussion concerning bus service. A significant cause of poor service in New York is congestion. A number of measures are capable of improving congestion and consequently, bus service. Due to the city’s limited capacity at implementing short-term solutions, the most highly problematic routes should receive priority. Routes with slow speeds, high rates of bunching and high ridership are concentrated in Manhattan and Downtown Brooklyn which also cater to the most subway riders. -

Volume 2: Main Report SCOTTISH PARLIAMENT – SESSION 2
EDINBURGH TRAM NETWORK EDINBURGH TRAM (LINE TWO) BILL Environmental Statement: Volume 2: Main Report SCOTTISH PARLIAMENT – SESSION 2 PREFACE The Edinburgh Tram Line 2 Environmental Statement is published in five volumes: • Volume 1 Non-Technical Summary • Volume 2 Environmental Statement: Main Report • Volume 3 Figures • Volume 4 Appendices to Main Report • Volume 5 Protected Species Report (Confidential) This document is Volume 2. Table of Contents VOLUME 2 ENVIRONMENTAL STATEMENT: MAIN REPORT ABBREVIATIONS 1 INTRODUCTION 1.1 Background 1-1 1.2 Tram Line 2 and the Tram Network 1-1 1.3 The Environmental Impact Assessment of Tram Line 2 1-1 1.4 The EIA Process 1-1 1.5 Relationship Between Tram Line 1 and Tram Line 2 1-2 1.6 Authors 1-2 1.7 Structure of ES 1-3 2 THE PROPOSED SCHEME 2.1 Introduction 2-1 2.2 The Need for the Scheme 2-1 2.3 Scheme Alternatives 2-2 2.4 Scheme Description 2-4 2.5 Tram Line 2 Infrastructure 2-7 2.6 The Construction Phase 2-11 2.7 Operation of Tram Line 2 2-14 3 APPROACH TO THE EIA 3.1 Introduction 3-1 3.2 Parliamentary Requirements and the EIA Regulations 3-1 3.3 The EIA Process 3-1 3.4 Approach to the Assessment of Impacts 3-2 3.5 Uncertainty, Assumptions and Limitations 3-4 3.6 Scope of the Environmental Statement and Consultation 3-6 4 POLICY CONTEXT 4.1 Introduction 4-1 4.2 Methods 4-1 4.3 National and Regional Planning Guidance 4-3 4.4 Planning Policies of The Local Authority 4-6 4.5 Summary 4-13 5 TRAFFIC AND TRANSPORT 5.1 Introduction 5-1 5.2 Methods 5-1 5.3 Baseline Situation 5-4 5.4 Construction Effects -

55-01 Myrtle Avenue Queens, NY Ridgewood
RETAIL/ RESTAURANT/ FITNESS/ MEDICAL 55-01 Myrtle Avenue 4,510 SF Queens, NY Available for Lease Ridgewood Located Between St. Nicholas Avenue and Madison Avenue Size Neighbors Comments Transportation 4,510 SF - Ground Floor GoHealth, Zwanger-Pesiri, Zwanger-Pesiri already signed 2017 Ridership Report 4,069 SF - Storage Basement Blink Fitness, Planet Fitness, Heart of Ridgewood CVS, Duane Reade, Starbucks, Myrtle-Wyckoff Avenue L M Asking Rent T-Mobile, Investor Bank, One Block from Subway Annual 7,302,376 Upon Request Bath & Boday Works, Food Weekday 22,434 New Construction Weekend 29,498 Bazzar, Auto Zone, Liberty Possession Department Store Walking distance from two Q3 2019 open air parking Food Bazzars Q55 55 Annual 2,217,379 Frontage Several new residential Weekday 7,147 43’ on Myrtle Avenue developments within a few Weekend 7,407 blocks from the site 16’+ on St. Nicholas B52 Corner location with tremendous 52 Annual 3,579,984 visibility Weekday 11,326 Weekend 15,620 Contact our exclusive agents: Q58 58 Annual 9,467,869 Andrew Clemens Ben Weiner Craig Gelberg Weekday 28,810 [email protected] [email protected] [email protected] Weekend 39,407 718.233.6565 718.233.6565 718.233.6565 Greenpoint Landing Newtown Barge Park Future Box St Park Greenpoint Playground DUPONT STREET COMMERCIAL ST EAGLE STREET Glasserie WEST STREET WEST Eagles Trading Company CLAY STREET Pure Barre FREEMAN STREET BOX STREET Fastenal Evil & Love Tattoo Church Achilles Heel DUPONT STREET Gabriel's Collision Corner Alluvium GREEN STREET New Supermarket Alameda -

NYCT Bus & MTA Bus Employee On-Duty Lost-Time Accident Rate
Bus Company Transit & Bus Committee Meeting June 2013 Committee Members M. Lebow, Chair F. Ferrer, Acting MTA Chairman J. Banks III, Vice Chair S. Metzger J. Sedore, Jr. M. Page J. Kay A. Albert C. Moerdler D. Paterson E. Watt A. Cappelli MEETING AGENDA NEW YORK CITY TRANSIT & BUS COMMITTEE June 3, 2013 - 10:30 AM 347 Madison Avenue Fifth Floor Board Room, New York, NY AGENDA ITEMS PUBLIC COMMENT PERIOD 1. APPROVAL OF MINUTES – APRIL 22, 2013 1.1 2. COMMITTEE WORK PLAN 2.1 3. OPERATIONS PERFORMANCE SUMMARY ¾ April Operations Report 3.1 ¾ March Operations Report 3.34 4. FINANCIAL REPORTS ¾ March NYCT Financial & Ridership Report 4.1 ¾ March SIR Financial & Ridership Report 4.23 ¾ March MTA Bus Financial & Ridership Report 4.34 ¾ April NYC Transit & MTA Bus Flash Reports (under separate cover) ¾ Capital Program Status Report 4.47 5. PROCUREMENTS 5.1 ¾ NYCT Non-Competitive 5.5 ¾ NYCT Competitive 5.6 ¾ MTACC Competitive 5.10 ¾ MTA Bus Competitive 5.11 ¾ NYCT Ratifications 5.12 ¾ MTACC Ratifications 5.13 6. SERVICE CHANGES ¾ NYCT Implement B67 Extension to Brooklyn Navy Yard (For Approval) 6.1 ¾ NYCT Implement New B32 Bus Service in Brooklyn and Queens (For Approval) 6.8 ¾ NYCT Reroute M100 Bus Service in East Harlem 6.15 ¾ NYCT Bus Schedule Changes, Effective September 2013 6.19 ¾ MTA Bus Implement New Q70 Limited Stop Service (For Approval) 6.24 ¾ MTA Bus Schedule Changes, Effective September 2013 6.35 7. SPECIAL REPORTS & PRESENTATIONS ¾ April MetroCard Report 7.1 ¾ March MetroCard Report 7.5 8. STANDARD FOLLOW-UP REPORTS ¾ Escalator & Elevator Service Report 8.1 ¾ Transit Adjudication Bureau Report 8.24 ¾ NYC Transit & MTA Bus EEO Report 8.26 9. -
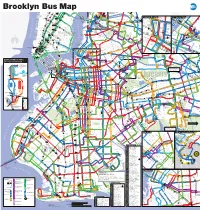
Brooklyn Bus Map
Brooklyn Bus Map 7 7 Queensboro Q M R Northern Blvd 23 St C E BM Plaza 0 N W R W 5 Q Court Sq Q 1 0 5 AV 6 1 2 New 3 23 St 1 28 St 4 5 103 69 Q 6 7 8 9 10 33 St 7 7 E 34 ST Q 66 37 AV 23 St F M Q18 to HIGH LINE Chelsea 44 DR 39 E M Astoria E M R Queens Plaza to BROADWAY Jersey W 14 ST QUEENS MIDTOWN Court Sq- Q104 ELEVATED 23 ST 7 23 St 39 AV Astoria Q 7 M R 65 St Q PARK 18 St 1 X 6 Q 18 FEDERAL 32 Q Jackson Hts Downtown Brooklyn LIC / Queens Plaza 102 Long 28 St Q Downtown Brooklyn LIC / Queens Plaza 27 MADISON AV E 28 ST Roosevelt Av BUILDING 67 14 St A C E TUNNEL 32 44 ST 58 ST L 8 Av Hunters 62 70 Q R R W 67 G 21 ST Q70 SBS 14 St X Q SKILLMAN AV E F 23 St E 34 St / VERNON BLVD 21 St G Court Sq to LaGuardia SBS F Island 66 THOMSO 48 ST F 28 Point 60 M R ED KOCH Woodside Q Q CADMAN PLAZA WEST Meatpacking District Midtown Vernon Blvd 35 ST Q LIRR TILLARY ST 14 St 40 ST E 1 2 3 M Jackson Av 7 JACKSONAV SUNNYSIDE ROTUNDA East River Ferry N AV 104 WOODSIDE 53 70 Q 40 AV HENRY ST N City 6 23 St YARD 43 AV Q 6 Av Hunters Point South / 7 46 St SBS SBS 3 GALLERY R L UNION 7 LT AV 2 QUEENSBORO BROADWAY LIRR Bliss St E BRIDGE W 69 Long Island City 69 St Q32 to PIERREPONT ST 21 ST V E 7 33 St 7 7 7 7 52 41 26 SQUARE HUNTERSPOINT AV WOOD 69 ST Q E 23 ST WATERSIDE East River Ferry Rawson St ROOSEV 61 St Jackson 74 St LIRR Q 49 AV Woodside 100 PARK PARK AV S 40 St 7 52 St Heights Bway Q I PLAZA LONG 7 7 SIDE 38 26 41 AV A 2 ST Hunters 67 Lowery St AV 54 57 WEST ST IRVING PL ISLAND CITY VAN DAM ST Sunnyside 103 Point Av 58 ST Q SOUTH 11 ST 6 3 AV 7 SEVENTH AV Q BROOKLYN 103 BORDEN AV BM 30 ST Q Q 25 L N Q R 27 ST Q 32 Q W 31 ST R 5 Peter QUEENS BLVD A Christopher St-Sheridan Sq 1 14 St S NEWTOWN CREEK 39 47 AV HISTORICAL ADAMS ST 14 St-Union Sq 5 40 ST 18 47 JAY ST 102 Roosevelt Union Sq 2 AV MONTAGUE ST 60 Q F 21 St-Queensbridge 4 Cooper McGUINNESS BLVD 48 AV SOCIETY JOHNSON ST THE AMERICAS 32 QUEENS PLAZA S. -
B47 Local Service
Bus Timetable Effective as of January 6, 2019 New York City Transit B47 Local Service a Between Kings Plaza and Bedford- Stuyvesant If you think your bus operator deserves an Apple Award — our special recognition for service, courtesy and professionalism — call 511 and give us the badge or bus number. Fares – MetroCard® is accepted for all MTA New York City trains (including Staten Island Railway - SIR), and, local, Limited-Stop and +SelectBusService buses (at MetroCard fare collection machines). Express buses only accept 7-Day Express Bus Plus MetroCard or Pay-Per-Ride MetroCard. All of our buses and +SelectBusService Coin Fare Collector machines accept exact fare in coins. Dollar bills, pennies, and half-dollar coins are not accepted. Free Transfers – Unlimited Ride MetroCard permits free transfers to all but our express buses (between subway and local bus, local bus and local bus etc.) Pay-Per-Ride MetroCard allows one free transfer of equal or lesser value if you complete your transfer within two hours of the time you pay your full fare with the same MetroCard. If you pay your local bus fare with coins, ask for a free electronic paper transfer to use on another local bus. Reduced-Fare Benefits – You are eligible for reduced-fare benefits if you are at least 65 years of age or have a qualifying disability. Benefits are available (except on peak-hour express buses) with proper identification, including Reduced-Fare MetroCard or Medicare card (Medicaid cards do not qualify). Children – The subway, SIR, local, Limited-Stop, and +SelectBusService buses permit up to three children, 44 inches tall and under to ride free when accompanied by an adult paying full fare. -

Bushwick BEDFORD-STUYVESANT • EAST WILLIAMSBURG • RIDGEWOOD
Bushwick BEDFORD-STUYVESANT • EAST WILLIAMSBURG • RIDGEWOOD Ellery St, E1, E2 Jefferson Av, G12, H11, L7, K9, L2 Morgan Av, A4, C4 Stanwix St, D3, F4 83rd Police Precinct, F9 Crossover Baptist Church, J1 JHS 162, B8 PS 25, J1 Ridgewood Salvation Army, D6 Universal Baptist Church, L6 Streets & Bridges Evergreen Av, D4, G7, J9 Jefferson St, C7, E5, F3, F4 Myrtle Av, F7, F9, G2 Starr St, A9, B8, C7, E6 Akwaaba Mansion Bed & Breakfast, M4 Davis Medical Building, F4 JHS 296, K11 PS 26, H4 Ridgewood/Bushwick Youth Center, F10 Upper Room Baptist Church, J2 Key accessible Dekalb Public Library, G5 Justice Gilbert Ramirez Playground, B3 entrance & exit Fairview Av, B12 Johnson Av, B4, B6, C1 Noll St, D4, E4 Stewart Av, B6 Antioch Baptist Church, J3 PS 44, L1 Robert F. Kennedy Memorial School, E1 Washington Irving Public Library, F11 Y W Arion Pl, F3 XP E S Fayette St, E2 Knickerbocker Av, C5, D7, G11 Onderdonk Av, C11 Stockholm St, B10, D8, E7 Asmbly of God Church of Bedford Stuyvesant, H1 East Williamsburg, B3 Knickerbocker United Methodist Church, F9 PS 45, L11 Roosevelt Houses, H2 Wayside Baptist Church, M11 N E E Bainbridge St, M9 U Q Flushing Av, B7, D5, E2 Kosciuszko St, H3, H5 Palmetto St, E11, H9, J8 Stockton St, F2 Beattie Square, G3 Eleanor Roosevelt Playground, D11 Kosciusko Street Garden, H2 PS 59 Sumner Playground, F2 Rose of Sharon Church of Christ, G4 Williamsburg Houses, A1 N Y L Beaver St, E3 K El Shabazz Playground, M5 O Forrest St, D4 Kossuth Pl, H6 Park Av, F1 Stuyvesant Av, G3, L4 Bedford District Health Center, L1 Lafayette